Co-reporter:Qian Wang, Hong Yan Zhao, Po-Kam Lo, William W. Y. Lam, Kai-Chung Lau, and Tai-Chu Lau
Inorganic Chemistry November 6, 2017 Volume 56(Issue 21) pp:12699-12699
Publication Date(Web):October 13, 2017
DOI:10.1021/acs.inorgchem.7b02509
We have previously reported that the oxidation of SO32– to SO42– by a trans-dioxoruthenium(VI) complex, [RuVI(TMC)(O)2)]2+ (RuVI; TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazcyclotetradecane) in aqueous solutions occurs via an O-atom transfer mechanism. In this work, we have reinvestigated the effects of the pH on the oxidation of SIV by RuVI in more detail in order to obtain kinetic data for the HSO3– pathway. The HSO3– pathway exhibits a deuterium isotope effect of 17.4, which indicates that O–H bond breaking occurs in the rate-limiting step. Density functional theory calculations have been performed that suggest that the oxidation of HSO3– by RuVI may occur via a concerted or stepwise proton-coupled O-atom transfer mechanism.
Co-reporter:Yi Pan;Zhihong Luo;Yih-Chung Chang;C. Y. Ng
The Journal of Physical Chemistry A January 26, 2017 Volume 121(Issue 3) pp:669-679
Publication Date(Web):December 2, 2016
DOI:10.1021/acs.jpca.6b09491
The ionization energies (IEs) of TiO and TiO2 and the 0 K bond dissociation energies (D0) and the heats of formation at 0 K (ΔH°f0) and 298 K (ΔH°f298) for TiO/TiO+ and TiO2/TiO2+ are predicted by the wave-function-based CCSDTQ/CBS approach. The CCSDTQ/CBS calculations involve the approximation to the complete basis set (CBS) limit at the coupled cluster level up to full quadruple excitations along with the zero-point vibrational energy (ZPVE), high-order correlation (HOC), core–valence (CV) electronic, spin–orbit (SO) coupling, and scalar relativistic (SR) effect corrections. The present calculations yield IE(TiO) = 6.815 eV and are in good agreement with the experimental IE value of 6.819 80 ± 0.000 10 eV determined in a two-color laser-pulsed field ionization-photoelectron (PFI-PE) study. The CCSDT and MRCI+Q methods give the best predictions to the harmonic frequencies: ωe (ωe+) = 1013 (1069) and 1027 (1059) cm–1 and the bond lengths re (re+) = 1.625 (1.587) and 1.621 (1.588) Å, for TiO (TiO+) compared with the experimental values. Two nearly degenerate, stable structures are found for TiO2 cation: TiO2+(C2v) structure has two equivalent TiO bonds, while the TiO2+(Cs) structure features a long and a short TiO bond. The IEs for the TiO2+(C2v)←TiO2 and TiO2+(Cs)←TiO2 ionization transitions are calculated to be 9.515 and 9.525 eV, respectively, giving the theoretical adiabatic IE value in good agreement with the experiment IE(TiO2) = 9.573 55 ± 0.000 15 eV obtained in the previous vacuum ultraviolet (VUV)–PFI-PE study of TiO2. The potential energy surface of TiO2+ along the normal vibrational coordinates of asymmetric stretching mode (ω3+) is nearly flat and exhibits a double-well potential with the well of TiO2+ (Cs) situated around the central well of TiO2+(C2v). This makes the theoretical calculation of ω3+ infeasible. For the symmetric stretching (ω1+), the current theoretical predictions overestimate the experimental value of 829.1 ± 2.0 cm–1 by more than 100 cm–1. This work together with the previous experimental and theoretical investigations supports the conclusion that the CCSDTQ/CBS approach is capable of providing reliable IE and D0 predictions for TiO/TiO+ and TiO2/TiO2+ with error limits less than or equal to 60 meV. The CCSDTQ/CBS calculations give the predictions of D0(Ti+–O) – D0(Ti–O) = 0.004 eV and D0(O–TiO) – D0(O–TiO+) = 2.699 eV, which are also consistent with the respective experimental determination of 0.008 32 ± 0.000 10 and 2.753 75 ± 0.000 18 eV.
Co-reporter:Dr. Hoi-Ki Kwong;Dr. Po-Kam Lo;Dr. Shek-Man Yiu;Dr. Hajime Hirao;Dr. Kai-Chung Lau; Tai-Chu Lau
Angewandte Chemie 2017 Volume 129(Issue 40) pp:12428-12431
Publication Date(Web):2017/09/25
DOI:10.1002/ange.201705986
AbstractThe OsVI nitrido complex, OsVI(N)(quin)2(OTs) (1, quin=2-quinaldinate, OTs=tosylate), is a highly selective and efficient catalyst for the ring hydroxylation of alkylbenzenes with H2O2 at room temperature. Oxidation of various alkylbenzenes occurs with ring/chain oxidation ratios ranging from 96.7/3.3 to 99.9/0.1, and total product yields from 93 % to 98 %. Moreover, turnover numbers up to 6360, 5670, and 3880 can be achieved for the oxidation of p-xylene, ethylbenzene, and mesitylene, respectively. Density functional theory calculations suggest that the active intermediate is an OsVIII nitrido oxo species.
Co-reporter:Dr. Hoi-Ki Kwong;Dr. Po-Kam Lo;Dr. Shek-Man Yiu;Dr. Hajime Hirao;Dr. Kai-Chung Lau; Tai-Chu Lau
Angewandte Chemie International Edition 2017 Volume 56(Issue 40) pp:12260-12263
Publication Date(Web):2017/09/25
DOI:10.1002/anie.201705986
AbstractThe OsVI nitrido complex, OsVI(N)(quin)2(OTs) (1, quin=2-quinaldinate, OTs=tosylate), is a highly selective and efficient catalyst for the ring hydroxylation of alkylbenzenes with H2O2 at room temperature. Oxidation of various alkylbenzenes occurs with ring/chain oxidation ratios ranging from 96.7/3.3 to 99.9/0.1, and total product yields from 93 % to 98 %. Moreover, turnover numbers up to 6360, 5670, and 3880 can be achieved for the oxidation of p-xylene, ethylbenzene, and mesitylene, respectively. Density functional theory calculations suggest that the active intermediate is an OsVIII nitrido oxo species.
Co-reporter:Ziyong Chen, Kai-Chung LauGustavo A. Garcia, Laurent NahonDušan K. Božanić, Lionel Poisson, Muneerah Mogren Al-Mogren, Martin SchwellJoseph S. Francisco, Ayad Bellili, Majdi Hochlaf
Journal of the American Chemical Society 2016 Volume 138(Issue 51) pp:16596-16599
Publication Date(Web):December 6, 2016
DOI:10.1021/jacs.6b10413
Co-reporter:A. Bellili, Y. Pan, M.M. Al Mogren, K.C. Lau, M. Hochlaf
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2016 Volume 164() pp:1-7
Publication Date(Web):5 July 2016
DOI:10.1016/j.saa.2016.03.043
•Accurate spectroscopic properties of 2-quinolinone cation•Adiabatic and vertical ionization energies of 2-quinolinone•Charge redistribution upon ionization and effect of annelation on 2-pyridoneUsing first principle methodologies, we characterize the lowest electronic states of 2-quinolinone+ cation. The ground state of this ion is of X˜2A″ nature. We deduce the adiabatic ionization energy of 2-quinolinone to be equal 8.249 eV using the explicitly correlated coupled cluster level and where zero point vibrational energy, core-valence and scalar relativistic effects are taken into account. We examine also the ionization induced structural changes and vibrational shifts and analyze the electron density differences between the neutral and ionic species. These data show that the formation of 2-quinolinone+X˜2A″ from 2-quinolinone affects strongly the HNCO group, whereas the carbon skeletal is perturbed when the upper electronic cationic states are populated. The comparison to 2-pyridone allows the elucidation of the effect of benzene ring fused with this heterocyclic ring. Since quinolones and pyridones are both model systems of DNA bases, these findings might help in understanding the charge redistribution in these biological entities upon ionization.
Co-reporter:Dr. Li Ma;Dr. William W. Y. Lam;Dr. Po-Kam Lo;Dr. Kai-Chung Lau; Tai-Chu Lau
Angewandte Chemie International Edition 2016 Volume 55( Issue 9) pp:3012-3016
Publication Date(Web):
DOI:10.1002/anie.201510156
Abstract
Although FeO42− (ferrate(IV)) is a very strong oxidant that readily oxidizes water in acidic medium, at pH 9–10 it is relatively stable (<2 % decomposition after 1 h at 298 K). However, FeO42− is readily activated by Ca2+ at pH 9–10 to generate O2. The reaction has the following rate law: d[O2]/dt=kCa[Ca2+][FeO42−]2. 18O-labeling experiments show that both O atoms in O2 come from FeO42−. These results together with DFT calculations suggest that the function of Ca2+ is to facilitate O–O coupling between two FeO42-ions by bridging them together. Similar activating effects are also observed with Mg2+ and Sr2+.
Co-reporter:Zhihong Luo, Yih-Chung Chang, Yi Pan, Kai-Chung Lau, and C. Y. Ng
The Journal of Physical Chemistry A 2016 Volume 120(Issue 27) pp:4643-4654
Publication Date(Web):September 24, 2015
DOI:10.1021/acs.jpca.5b07939
By employing the two-color visible–ultraviolet (vis–UV) laser pulsed field ionization–photoelectron (PFI–PE) measurement, we have obtained rotationally selected and resolved photoelectron spectra for the MoO+(X4Σ–; v+ = 0, 1, and 2) and MoO+(a2Δ3/2,5/2; v+ = 0 and 1) cationic states. The unambiguous rotational assignments have made possible the determination of highly precise values for the band origin v00+ = 60 147.9 ± 0.8 cm–1, rotation constant B0+ = 0.4546 ± 0.0006 cm–1, spin–spin coupling constant λ = 26.454 ± 0.017 cm–1, and bond length re+ = 1.642 ± 0.001 Å for the MoO+(X4Σ–) ground state; v00+ = 60 556.4 ± 0.8 cm–1, B0+ = 0.4711 ± 0.0005 cm–1, and r0+ = 1.613 ± 0.001 Å for the MoO+ (a2Δ3/2) excited state; and v00+ = 61 718.2 ± 0.8 cm–1, B0+ = 0.4695 ± 0.0006 cm–1, and r0+ = 1.616 ± 0.001 Å for the MoO+ (a2Δ5/2) excited state. The ionization energy (IE) for MoO is determined to be IE(MoO) = 60 095.1 ± 0.8 cm–1 [7.4508 ± 0.0001 eV]. Furthermore, the vibrational constants are determined as ωe+ = 1000 ± 9 cm–1 and ωe+xe+ = 5 ± 3 cm–1 for MoO+(X4Σ–); the vibration spacing ΔG(1/2) for MoO+(a2Δ3/2) is also measured as 1065 ± 4 cm–1. On the basis of the thermochemical cycle, together with the known IE(Mo) and the IE(MoO) determined in this study, the difference of 0 K bond dissociation energy for MoO+ and that for MoO is determined to be D0(Mo+–O) – D0(Mo–O) = IE(Mo) – IE(MoO) = −2890.8 ± 0.9 cm–1 [−0.3584 ± 0.0001 eV]. The energetic and spectroscopic values determined here have been used for benchmarking calculations at the CCSDTQ/CBS level of theory. The CCSDTQ/CBS predictions, IE(MoO) = 7.457 eV, re+ = 1.651 Å, ωe+ = 974 cm–1, D0(Mo–O) = 5.386, and D0(Mo+–O) = 5.015 eV, are found to be in good agreement with the vis–UV PFI–PE measurements. We also recommend a set of equally reliable CCSDTQ/CBS thermochemical values for MoO and MoO+: ΔH°f0(MoO) = 383.7, ΔH°f298(MoO) = 384.0, ΔH°f0(MoO+) = 1103.2, and ΔH°f298(MoO+) = 1103.5 kJ mol–1.
Co-reporter:Dr. Qian Wang;Hong Yan Zhao;Dr. Wai-Lun Man;Dr. William W. Y. Lam;Dr. Kai-Chung Lau; Tai-Chu Lau
Chemistry - A European Journal 2016 Volume 22( Issue 31) pp:10754-10758
Publication Date(Web):
DOI:10.1002/chem.201601848
Abstract
The kinetics and mechanism of the reaction of SIV (SO32−+HSO3−) with a ruthenium(VI) nitrido complex, [(L)RuVI(N)(OH2)]+ (RuVIN, L=N,N′-bis(salicylidene)-o-cyclohexyldiamine dianion), in aqueous acidic solutions are reported. The kinetic results are consistent with parallel pathways involving oxidation of HSO3− and SO32− by RuVIN. A deuterium isotope effect of 4.7 is observed in the HSO3− pathway. Based on experimental results and DFT calculations the proposed mechanism involves concerted N−S bond formation (partial N-atom transfer) between RuVIN and HSO3− and H+ transfer from HSO3− to a H2O molecule.
Co-reporter:Dr. Li Ma;Dr. William W. Y. Lam;Dr. Po-Kam Lo;Dr. Kai-Chung Lau; Tai-Chu Lau
Angewandte Chemie International Edition 2016 Volume 55( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/anie.201680961
Co-reporter:Dr. Li Ma;Dr. William W. Y. Lam;Dr. Po-Kam Lo;Dr. Kai-Chung Lau; Tai-Chu Lau
Angewandte Chemie 2016 Volume 128( Issue 9) pp:3064-3068
Publication Date(Web):
DOI:10.1002/ange.201510156
Abstract
Although FeO42− (ferrate(IV)) is a very strong oxidant that readily oxidizes water in acidic medium, at pH 9–10 it is relatively stable (<2 % decomposition after 1 h at 298 K). However, FeO42− is readily activated by Ca2+ at pH 9–10 to generate O2. The reaction has the following rate law: d[O2]/dt=kCa[Ca2+][FeO42−]2. 18O-labeling experiments show that both O atoms in O2 come from FeO42−. These results together with DFT calculations suggest that the function of Ca2+ is to facilitate O–O coupling between two FeO42-ions by bridging them together. Similar activating effects are also observed with Mg2+ and Sr2+.
Co-reporter:Dr. Li Ma;Dr. William W. Y. Lam;Dr. Po-Kam Lo;Dr. Kai-Chung Lau; Tai-Chu Lau
Angewandte Chemie 2016 Volume 128( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/ange.201680961
Co-reporter:Yih Chung Chang, Zhihong Luo, Yi Pan, Zheng Zhang, Ying-Nan Song, Sophie Yajin Kuang, Qing Zhu Yin, Kai-Chung Lau and C. Y. Ng
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 15) pp:9780-9793
Publication Date(Web):26 Feb 2015
DOI:10.1039/C5CP00371G
By employing two-color visible (VIS)-ultraviolet (UV) laser photoionization and pulsed field ionization-photoelectron (PFI-PE) techniques, we have obtained highly rotationally resolved photoelectron spectra for vanadium monocarbide cations (VC+). The state-to-state VIS-UV-PFI-PE spectra thus obtained allow unambiguous assignments for the photoionization rotational transitions, resulting in a highly precise value for the adiabatic ionization energy (IE) of vanadium monocarbide (VC), IE(VC) = 57512.0 ± 0.8 cm−1 (7.13058 ± 0.00010 eV), which is defined as the energy of the VC+(X3Δ1; v+ = 0; J+ = 1) ← VC(X2Δ3/2; v′′ = 0; J′′ = 3/2) photoionization transition. The spectroscopic constants for VC+(X3Δ1) determined in the present study include the harmonic vibrational frequency ωe+ = 896.4 ± 0.8 cm−1, the anharmonicity constant ωe+xe+ = 5.7 ± 0.8 cm−1, the rotational constants Be+ = 0.6338 ± 0.0025 cm−1 and αe+ = 0.0033 ± 0.0007 cm−1, the equilibrium bond length re+ = 1.6549 ± 0.0003 Å, and the spin–orbit coupling constant A = 75.2 ± 0.8 cm−1 for VC+(X3Δ1,2,3). These highly precise energetic and spectroscopic data are used to benchmark state-of-the-art CCSDTQ/CBS calculations. In general, good agreement is found between the theoretical predictions and experimental results. The theoretical calculations yield the values, IE(VC) = 7.126 eV; the 0 K bond dissociation energies: D0(V–C) = 4.023 eV and D0(V+–C) = 3.663 eV; and heats of formation: , , , and kJ mol−1.
Co-reporter:Wing-Kin Chu;Xi-Guang Wei;Dr. Shek-Man Yiu;Dr. Chi-Chiu Ko;Dr. Kai-Chung Lau
Chemistry - A European Journal 2015 Volume 21( Issue 6) pp:2603-2612
Publication Date(Web):
DOI:10.1002/chem.201405291
Abstract
A new series of neutral isocyanoborato rhenium(I) diimine complexes [Re(CO)3(N^N)(CNBR3)], where N^N=bpy, 4,4′-Me2bpy, phen, 4,7-Me2phen, 2,9-Me2phen, 3,4,7,8-Me4phen; R=C6F5, C6H5, Cl, 4-ClC6H4, 3,5-(CF3)2C6H3, with various isocyanoborate and diimine ligands of diverse electronic and steric nature have been synthesized and characterized. The X-ray crystal structures of six complexes have also been determined. These complexes displayed intense bluish green to yellow phosphorescence at room temperature in dichloromethane solution. The photophysical and electrochemical properties of these complexes had been investigated. To elucidate the electronic structures and transitions of these complexes, DFT and TD-DFT calculations have been performed, which revealed that the lowest-energy electronic transition associated with these complexes originates from a mixture of MLCT [dπ(Re)π*(N^N)] and LLCT [π(CNBR3)π*(N^N)] transitions.
Co-reporter:Youssef Majdi, Majdi Hochlaf , Yi Pan, Kai-Chung Lau , Lionel Poisson , Gustavo A. Garcia, Laurent Nahon , Muneerah Mogren Al-Mogren and Martin Schwell
The Journal of Physical Chemistry A 2015 Volume 119(Issue 23) pp:5951-5958
Publication Date(Web):December 24, 2014
DOI:10.1021/jp510716c
We report on the vibronic structure of the ground state X̃2A″ of the thymine cation, which has been measured using a threshold photoelectron photoion coincidence technique and vacuum ultraviolet synchrotron radiation. The threshold photoelectron spectrum, recorded over ∼0.7 eV above the ionization potential (i.e., covering the whole ground state of the cation) shows rich vibrational structure that has been assigned with the help of calculated anharmonic modes of the ground electronic cation state at the PBE0/aug-cc-pVDZ level of theory. The adiabatic ionization energy has been experimentally determined as AIE = 8.913 ± 0.005 eV, in very good agreement with previous high resolution results. The corresponding theoretical value of AIE = 8.917 eV has been calculated in this work with the explicitly correlated method/basis set (R)CCSD(T)-F12/cc-pVTZ-F12, which validates the theoretical approach and benchmarks its accuracy for future studies of medium-sized biological molecules.
Co-reporter:Majdi Hochlaf, Yi Pan and Kai-Chung Lau , Youssef Majdi, Lionel Poisson , Gustavo A. Garcia, Laurent Nahon , Muneerah Mogren Al Mogren and Martin Schwell
The Journal of Physical Chemistry A 2015 Volume 119(Issue 7) pp:1146-1153
Publication Date(Web):January 22, 2015
DOI:10.1021/acs.jpca.5b00466
For fully understanding the light–molecule interaction dynamics at short time scales, recent theoretical and experimental studies proved the importance of accurate characterizations not only of the ground (D0) but also of the electronic excited states (e.g., D1) of molecules. While ground state investigations are currently straightforward, those of electronic excited states are not. Here, we characterized the à electronic state of ionic thymine (T+) DNA base using explicitly correlated coupled cluster ab initio methods and state-of-the-art synchrotron-based electron/ion coincidence techniques. The experimental spectrum is composed of rich and long vibrational progressions corresponding to the population of the low frequency modes of T+(Ã). This work challenges previous numerous works carried out on DNA bases using common synchrotron and VUV-based photoelectron spectroscopies. We provide hence a powerful theoretical and experimental framework to study the electronic structure of ionized DNA bases that could be generalized to other medium-sized biologically relevant systems.
Co-reporter:Kwan-Ming Ng
The Journal of Physical Chemistry C 2015 Volume 119(Issue 41) pp:23708-23720
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.jpcc.5b05957
Fundamental factors governing the ion-desorption efficiency and extent of internal-energy transfer to a chemical thermometer, benzylpyridinium ion ([BP]+), generated in the surface-assisted laser desorption/ionization (SALDI) process, were systematically investigated using noble metal nanoparticles (NPs), including AuNPs, AgNPs, PdNPs, and PtNPs, as substrates, with an average particle size of 1.7–3.1 nm in diameter. In the correlation of ion-desorption efficiency and internal-energy transfer with physicochemical properties of the NPs, laser-induced heating of the NPs, which are dependent on their photoabsorption efficiencies, was found to be a key factor in governing the ion-desorption efficiency and the extent of internal-energy transfer. This suggested that the thermal-driven desorption played a significant role in the ion-desorption process. In addition, a stronger binding affinity of [BP]+ to the surface of the NPs could hinder its desorption from the NPs, and this could be another factor in determining the ion-desorption efficiency. Moreover, metal NPs with lower melting points could also facilitate the ion-desorption process via the phase-transition process, which could lower the activation barrier (ΔG#) of the ion-desorption process by increasing the entropic change (ΔS#). The study reveals that high photoabsorption efficiency, weak binding interaction with analyte molecule, and low melting point could be critical for the design of SALDI substrates with efficient ion desorption.
Co-reporter:Li Ma ; Yi Pan ; Wai-Lun Man ; Hoi-Ki Kwong ; William W. Y. Lam ; Gui Chen ; Kai-Chung Lau ;Tai-Chu Lau
Journal of the American Chemical Society 2014 Volume 136(Issue 21) pp:7680-7687
Publication Date(Web):May 5, 2014
DOI:10.1021/ja5019546
The oxidation of various alkanes catalyzed by [MnV(N)(CN)4]2– using various terminal oxidants at room temperature has been investigated. Excellent yields of alcohols and ketones (>95%) are obtained using H2O2 as oxidant and CF3CH2OH as solvent. Good yields (>80%) are also obtained using (NH4)2[Ce(NO3)6] in CF3CH2OH/H2O. Kinetic isotope effects (KIEs) are determined by using an equimolar mixture of cyclohexane (c-C6H12) and cyclohexane-d12 (c-C6D12) as substrate. The KIEs are 3.1 ± 0.3 and 3.6 ± 0.2 for oxidation by H2O2 and Ce(IV), respectively. On the other hand, the rate constants for the formation of products using c-C6H12 or c-C6D12 as single substrate are the same. These results are consistent with initial rate-limiting formation of an active intermediate between [Mn(N)(CN)4]2– and H2O2 or CeIV, followed by H-atom abstraction from cyclohexane by the active intermediate. When PhCH2C(CH3)2OOH (MPPH) is used as oxidant for the oxidation of c-C6H12, the major products are c-C6H11OH, c-C6H10O, and PhCH2C(CH3)2OH (MPPOH), suggesting heterolytic cleavage of MPPH to generate a Mn═O intermediate. In the reaction of H2O2 with [Mn(N)(CN)4]2– in CF3CH2OH, a peak at m/z 628.1 was observed in the electrospray ionization mass spectrometry, which is assigned to the solvated manganese nitrido oxo species, (PPh4)[Mn(N)(O)(CN)4]−·CF3CH2OH. On the basis of the experimental results the proposed mechanism for catalytic alkane oxidation by [MnV(N)(CN)4]2–/ROOH involves initial rate-limiting O-atom transfer from ROOH to [Mn(N)(CN)4]2– to generate a manganese(VII) nitrido oxo active species, [MnVII(N)(O)(CN)4]2–, which then oxidizes alkanes (R′H) via a H-atom abstraction/O-rebound mechanism. The proposed mechanism is also supported by density functional theory calculations.
Co-reporter:Yi Pan, Kai-Chung Lau, Muneerah Mogren Al-Mogren, Ahmed Mahjoub, Majdi Hochlaf
Chemical Physics Letters 2014 Volume 613() pp:29-33
Publication Date(Web):3 October 2014
DOI:10.1016/j.cplett.2014.08.033
•Theoretical study on geometrical parameters and vibrational frequencies of neutral 2-quinolinol.•DFT and CCSD(T)-F12 energetics on lactam–lactim tautomarization and the cis–trans isomerization of the lactim.•The lactam form is confirmed to be the most stable isomer of 2-quinolinol.•Multiconfigurational calculations on electronic excitation of the S1 ← S0 transition.We treat theoretically 2-quinolinol(lactam), an analog of carbostyril and DNA bases. We characterized the ground state structure of 2-quinolinol and its isomer(lactim) using density functional theory(DFT). The reaction profile and energetics for lactam–lactim tautomerization and cis-lactim to trans-lactim isomerization predicted with explicitly correlated methods. We explored the pattern of the lowest singlet and triplet manifolds of states and electronic S1 ← S0 transitions using multiconfigurational methodologies. The theoretical results are compared with available experimental data and used to interpret the on-going photoelectron study of 2-quinolinol. Our analysis should help to understand the effect of tautomerism and aromaticity on the DNA bases.
Co-reporter:Po-Kam Lo and Kai-Chung Lau
The Journal of Physical Chemistry A 2014 Volume 118(Issue 13) pp:2498-2507
Publication Date(Web):March 12, 2014
DOI:10.1021/jp412323j
The ionization energy (IE), electron affinity (EA), and heats of formation (ΔH°f0/ΔH°f298) for cyclopentadienyl radical, cation, and anion, C5H5/C5H5+/C5H5–, have been calculated by wave function-based ab initio CCSDT/CBS approach, which involves approximation to complete basis set (CBS) limit at coupled-cluster level with up to full triple excitations (CCSDT). The zero-point vibrational energy correction, core–valence electronic correction, scalar relativistic effect, and higher-order corrections beyond the CCSD(T) wave function are included in these calculations. The allylic [C5H5(2A2)] and dienylic [C5H5(2B1)] forms of cyclopentadienyl radical are considered: the ground state structure exists in the dienyl form and it is about 30 meV more stable than the allylic structure. Both structures are lying closely and are interconvertible along the normal mode of b2 in-plane vibration. The CCSDT/CBS predictions (in eV) for IE[C5H5+(3A1′)←C5H5(2B1)] = 8.443, IE[C5H5+(1A1)←C5H5(2B1)] = 8.634 and EA[C5H5–(1A1′)←C5H5(2B1)] = 1.785 are consistent with the respective experimental values of 8.4268 ± 0.0005, 8.6170 ± 0.0005, and 1.808 ± 0.006, obtained from photoelectron spectroscopic measurements. The ΔH°f0/ΔH°f298’s (in kJ/mol) for C5H5/C5H5+/C5H5– have also been predicted by the CCSDT/CBS method: ΔH°f0/ΔH°f298[C5H5(2B1)] = 283.6/272.0, ΔH°f0/ΔH°f298[C5H5+(3A1′)] = 1098.2/1086.9, ΔH°f0/ΔH°f298[C5H5+(1A1)] = 1116.6/1106.0, and ΔH°f0/ΔH°f298[C5H5–(1A1′)] = 111.4/100.0. The comparisons between the CCSDT/CBS predictions and the experimental values suggest that the CCSDT/CBS procedure is capable of predicting reliable IE(C5H5)’s and EA(C5H5) with uncertainties of ±17 and ±23 meV, respectively.
Co-reporter:Wai-Lun Man ; Jianhui Xie ; Yi Pan ; William W. Y. Lam ; Hoi-Ki Kwong ; Kwok-Wa Ip ; Shek-Man Yiu ; Kai-Chung Lau ;Tai-Chu Lau
Journal of the American Chemical Society 2013 Volume 135(Issue 15) pp:5533-5536
Publication Date(Web):March 28, 2013
DOI:10.1021/ja401553d
We report experimental and computational studies of the facile oxidative C–N bond cleavage of anilines by a (salen)ruthenium(VI) nitrido complex. We provide evidence that the initial step involves nucleophilic attack of aniline at the nitrido ligand of the ruthenium complex, which is followed by proton and electron transfer to afford a (salen)ruthenium(II) diazonium intermediate. This intermediate then undergoes unimolecular decomposition to generate benzene and N2.
Co-reporter:Jerry C. Y. Lo, Michael C. W. Chan, Po-Kam Lo, Kai-Chung Lau, Takashi Ochiai, and Haruyuki Makio
Organometallics 2013 Volume 32(Issue 2) pp:449-459
Publication Date(Web):January 10, 2013
DOI:10.1021/om300832q
A series of Ti(IV) post-metallocene bis(benzyl) precatalysts supported by tridentate pyridine-2-phenolate-6-(σ-aryl) [O,N,C] ligands, featuring various substituents on the σ-aryl (directly adjacent to the pyridine ring: fluoro, trifluoromethyl, benzo [C4H4]) and phenolate groups (tert-butyl, trifluoromethyl, cumyl, 1,1-diphenylethyl), have been prepared. Multinuclear (including 1H, 13C, and 19F) NMR characterizations of the complexes have been performed. The principal purpose of this study was to investigate the impact of these substituents upon ethylene polymerization reactivity and polymer properties. The cumyl-phenolate σ-naphthyl Ti precatalyst, in conjunction with [Ph3C][B(C6F5)4], displays good activity and produces polyethylene with exceptionally high MW (Mn = 4 × 106) and an Mw/Mn value (2.5) approaching single-site character at 50 °C, but multisite behavior is apparent for other catalysts. DFT calculations have been performed to probe the polymerization behavior and the role of the py-adjacent substituent. These studies revealed the possibility of two distinct polymerization reactions, namely conventional and ethylene-assimilated (comprising initial ethylene insertion into the Ti–C(σ-aryl) bond) chain propagation, and found that the latter is kinetically preferred. Furthermore, the viability of another kinetically competitive pathway, namely the isomerization of the ethylene-assimilated [Ti−CH2CH2−aryl] species via β-H elimination and 2,1-reinsertion, was also indicated.
Co-reporter:Xin Wang , Kai-Chung Lau
The Journal of Physical Chemistry C 2012 Volume 116(Issue 43) pp:22749-22758
Publication Date(Web):October 9, 2012
DOI:10.1021/jp309226z
The charge-transfer properties of three diazapentacene derivatives, including 5,7,12,14-tetrachloro-6,13-diazapentacene (TCDAP), 5,7,12,14-tetrachloro-6,13-diaza-6,13-dihydropentacene (TCDAHP), and 5,7,12,14-tetrafluoro-6,13-diazapentacene (TFDAP), have been studied using density functional theory. The performance of five pure GGA and seven hybrid GGA functionals and G3MP2B3 method on the reorganization energy (λ) and mobility (μ) predictions of TCDAP has been examined. Both the B3LYP functional and the G3MP2B3 method give reliable predictions for the λ value. Using the reorganization energy at the G3MP2B3 level together with the transfer integral by BHandH, BHandHLYP, and M06-2X functionals yields electron mobilities of 3.44, 3.32, and 3.29 cm2 V–1 S–1 for TCDAP, respectively, which come fortuitously close to the experimental value of 3.39 cm2 V–1 S–1. Other density functionals also give mobility predictions in agreement with the experimental value to a factor of ∼2. The TCDAHP, a −NH derivative of TCDAP, is predicted to have a large hole and electron mobility of 2.30 and 3.89 cm2 V–1 S–1, respectively. Our results suggest that TCDAP is an n-channel material, while TCDAHP is an ambipolar organic semiconductor with simultaneous hole and electron transport properties. By the substitution of chlorine with fluorine in TCDAP, we find that TFDAP is very similar to TCDAP in terms of the molecular and crystal structure and HOMO/LUMO property. TFDAP is an n-type semiconductor but with a larger electron mobility of 3.51 cm2 V–1 S–1. All theoretical predictions are based on the crystal structures obtained with PBC model and B97D functional. The transfer integral calculations along the four dominant hopping pathways reveal that the hole and electron transport processes occur via the parallel routes between two neighboring molecules with π-stacking interactions. On the basis of the angular resolution anisotropic mobility analyses, TCDAP, TCDAHP, and TFDAP show remarkably different anisotropic behaviors in comparison with the 6,13-dihydro-6,13-diazapentacene (DHDAP).
Co-reporter:Shek-Man Yiu, Wai-Lun Man, Xin Wang, William W. Y. Lam, Siu-Mui Ng, Hoi-Ki Kwong, Kai-Chung Lau and Tai-Chu Lau
Chemical Communications 2011 vol. 47(Issue 14) pp:4159-4161
Publication Date(Web):28 Feb 2011
DOI:10.1039/C1CC00019E
MnO4− is activated by BF3 to undergo intramolecular coupling of two oxo ligands to generate O2. DFT calculations suggest that there should be a spin intercrossing between the singlet and triplet potential energy surfaces on going from the active intermediate [MnO2(OBF3)2]− to the O⋯O coupling transition state.
Co-reporter:Hoi-Ki Kwong, Po-Kam Lo, Kai-Chung Lau and Tai-Chu Lau
Chemical Communications 2011 vol. 47(Issue 14) pp:4273-4275
Publication Date(Web):07 Mar 2011
DOI:10.1039/C0CC05487A
The manganese(V) nitrido complex (PPh4)2[Mn(N)(CN)4] is an active catalyst for alkene epoxidation and alcohol oxidation using H2O2 as an oxidant. The catalytic oxidation is greatly enhanced by the addition of just one equivalent of acetic acid. The oxidation of ethene by this system has been studied computationally by the DFT method.
Co-reporter:Hongxia Du, Po-Kam Lo, Zongmin Hu, Haojun Liang, Kai-Chung Lau, Yi-Ning Wang, William W. Y. Lam and Tai-Chu Lau
Chemical Communications 2011 vol. 47(Issue 25) pp:7143-7145
Publication Date(Web):25 May 2011
DOI:10.1039/C1CC12024G
The oxidation of alcohols by KMnO4 is greatly accelerated by various Lewis acids. Notably the rate is increased by 4 orders of magnitude in the presence of Ca2+. The mechanisms of the oxidation of CH3OH and PhCH(OH)CH3 by MnO4− and BF3·MnO4− have also been studied computationally by the DFT method.
High-Level ab Initio Predictions for the Ionization Energies and Heats of Formation of Five-Membered-Ring Molecules: Thiophene, Furan, Pyrrole, 1,3-Cyclopentadiene, and Borole, C4H4X/C4H4X+ (X = S, O, NH, CH2, and BH)
Co-reporter:Po-Kam Lo and Kai-Chung Lau
The Journal of Physical Chemistry A 2011 Volume 115(Issue 5) pp:932-939
Publication Date(Web):January 6, 2011
DOI:10.1021/jp110499c
The ionization energies (IEs) and heats of formation (ΔH°f0/ΔH°f298) for thiophene (C4H4S), furan (C4H4O), pyrrole (C4H4NH), 1,3-cyclopentadiene (C4H4CH2), and borole (C4H4BH) have been calculated by the wave function-based ab initio CCSD(T)/CBS approach, which involves the approximation to the complete basis set (CBS) limit at the coupled-cluster level with single and double excitations plus a quasi-perturbative triple excitation [CCSD(T)]. Where appropriate, the zero-point vibrational energy correction (ZPVE), the core−valence electronic correction (CV), and the scalar relativistic effect (SR) are included in these calculations. The respective CCSD(T)/CBS predictions for C4H4S, C4H4O, C4H4NH, and C4H4CH2, being 8.888, 8.897, 8.222, and 8.582 eV, are in excellent agreement with the experimental values obtained from previous photoelectron and photoion measurements. The ΔH°f0/ΔH°f298 values for the aforementioned molecules and their corresponding cations have also been predicted by the CCSD(T)/CBS method, and the results are compared with the available experimental data. The comparisons between the CCSD(T)/CBS predictions and the experimental values for C4H4S, C4H4O, C4H4NH, and C4H4CH2 suggest that the CCSD(T)/CBS procedure is capable of predicting reliable IE values for five-membered-ring molecules with an uncertainty of ±13 meV. In view of the excellent agreements between the CCSD(T)/CBS predictions and the experimental values for C4H4S, C4H4O, C4H4NH, and C4H4CH2, the similar CCSD(T)/CBS IE and ΔH°f0/ΔH°f298 predictions for C4H4BH, whose thermochemical data are not readily available due to its reactive nature, should constitute a reliable data set. The CCSD(T)/CBS IE(C4H4BH) value is 8.868 eV, and ΔH°f0/ΔH°f298 values for C4H4BH and C4H4BH+ are 269.5/258.6 and 1125.1/1114.6 kJ/mol, respectively. The highest occupied molecular orbitals (HOMO) of C4H4S, C4H4O, C4H4NH, C4H4CH2, and C4H4BH have also been studied by the natural bond orbital (NBO) method, and the extent of π-electron delocalization in these five-membered rings are discussed in correlation with their molecular structures and orbitals.
Co-reporter:Xin Wang and Kai-Chung Lau and Wai-Kee Li
The Journal of Physical Chemistry A 2011 Volume 115(Issue 26) pp:7656-7663
Publication Date(Web):June 14, 2011
DOI:10.1021/jp200032a
Doping effects on the structural and electronic properties of ladderanes and ladder polysilanes have been studied using density functional theory. Two types of doping: substitution with isoelectronic atoms or heteroatoms (or radicals), have been used to design low band gap ladderanes. It is found that the B-doped [n]-ladderanes and 1,2 P-doped [n]-silaladderanes exhibit a very noticeable bent conformation, whereas the 1,2 and 1,3 N-doped ladderanes, P-doped ladderanes, and silaladderanes keep the relatively straight ladder shapes. The isoelectronic atom doping reduces the HOMO–LUMO (H-L) gaps of [n]-ladderanes but increases those of [n]-silaladderanes with n > 5. The present results show that isoelectronic atom doping is not an effective way to decrease the H-L gaps of ladderanes and silaladderanes. Heteroatom doping has a more pronounced effect than the isoelectronic atom doping. The HOMOs of heteroatom-doped ladderanes and silaladderanes are destabilized and LUMOs are stabilized, leading to significant reduction of H-L gaps. Most of the B-, N-, and P-doped [n]-silaladderanes we designed have H-L gaps below 1.5 eV. Therefore, it is expected that these silaladderanes are promising candidates of conductive or semiconductive materials. The heteroatom doping is a viable approach to reduce H-L gaps for the silaladderanes. In addition, it is found that nine different density functionals, including B3LYP, SVWN LDA, four pure GGAs, and three hybrid GGAs, as well as the time-dependent B3LYP method, all lead to the same predictions on the H-L gaps of ladderanes, silaladderanes, as well as their doped derivatives.
Co-reporter:Xin Wang and Kai-Chung Lau, Wai-Kee Li
The Journal of Physical Chemistry A 2009 Volume 113(Issue 14) pp:3413-3419
Publication Date(Web):March 18, 2009
DOI:10.1021/jp900161s
An investigation has been undertaken to study the large closed ladderanes C24H24 and their analogs. Thirteen isoelectronic species have been identified as local minima on the MP2(FU)/6-31G(d) potential energy surface, including three C24H24, four Si24H24, two N24, and four C24−xH24−xNx (x = 6, 8, 12) isomers. Of these 13 species, 11 are reported for the first time. Their structures, stabilities, HOMO−LUMO gaps, and G3(MP2) heats of formations are computationally obtained and discussed. These results are also compared with those of the related species already available in the literature. Our results show that molecules with 12-membered rings are highly energetic and much less stable than their corresponding isomers that do not have such rings. Isomer II of Si24H24 with anticonformations has a very small HOMO−LUMO gap of 2.56 eV, approaching the semiconductor range. Therefore, it is a candidate of potential semiconductor materials. Meanwhile, the N24 and other nitrogen-containing species are candidates for high-energy density materials. Our results also indicate that C18H18N6 and C16H16N8 may be good hexa- and octadentate ligands for metal cations.
Co-reporter:Lidong Zhang, Yang Pan, Huijun Guo, Taichang Zhang, Liusi Sheng and Fei Qi, Po-Kam Lo and Kai-Chung Lau
The Journal of Physical Chemistry A 2009 Volume 113(Issue 20) pp:5838-5845
Publication Date(Web):April 28, 2009
DOI:10.1021/jp9002565
We report a photoionization and dissociative photoionization study of β-alanine using IR laser desorption combined with synchrotron vacuum ultraviolet (VUV) photoionization mass spectrometry. Fragments at m/z = 45, 44, 43, and 30 yielded from photoionization are assigned to NH3CH2CH2+, NH2CHCH3+, NH2CHCH2+, and NH2CH2+, respectively. Some new conformation-specific dissociation channels and corresponding dissociation energies for the observed fragments are established and determined with the help of ab initio G3B3 calculations and measurements of photoionization efficiency (PIE) spectra. The theoretical values are in fair agreement with the experimental results. Three low-lying conformers of the β-alanine cation, including two gauche conformers G1+, G2+ and one anti conformer A+ are investigated by G3B3 calculations. The conformer G1+ (intramolecular hydrogen bonding N−H···O═C) is found to be another precursor in forming the NH3CH2CH2+ ion, which is complementary to the previously reported formation pathway that only occurs with the conformer G2+ (intramolecular hydrogen bonding O−H···N). Species NH2CHCH2+ may come from the contributions of G1+, G2+, and A+ via different dissociation pathways. The most abundant fragment ion, NH2CH2+, is formed from a direct C−C bond cleavage. Intramolecular hydrogen transfer processes dominate most of the fragmentation pathways of the β-alanine cation.
Co-reporter:Kai-Chung Lau, Yih-Chung Chang, Chow-Shing Lam and C. Y. Ng
The Journal of Physical Chemistry A 2009 Volume 113(Issue 52) pp:14321-14328
Publication Date(Web):September 23, 2009
DOI:10.1021/jp903218h
The ionization energy (IE) of FeC and the 0 K bond dissociation energies (D0) and the heats of formation at 0 K (ΔH°f0) and 298 K (ΔH°f298) for FeC and FeC+ are predicted by the single-reference wave function based CCSDTQ(Full)/CBS approach, which involves the approximation to the complete basis set (CBS) limit at the coupled cluster level up to full quadruple excitations. The zero-point vibrational energy (ZPVE) correction, the core−valence electronic corrections (up to CCSDT level), spin−orbit couplings, and relativistic effects (up to CCSDTQ level) are included in the calculations. The present calculations provide the correct symmetry predictions for the ground states of FeC and FeC+ to be 3Δ and 2Δ, respectively. We have also examined the theoretical harmonic vibrational frequencies of FeC/FeC+ at the ROHF-UCCSD(T) and UHF-UCCSD(T) levels. While the UHF-UCCSD(T) harmonic frequencies are in good agreement with the experimental measurements, the ROHF-UCCSD(T) yields significantly higher harmonic frequency predictions for FeC/FeC+. The CCSDTQ(Full)/CBS IE(FeC) = 7.565 eV is found to compare favorably with the experimental IE value of 7.59318 ± 0.00006 eV, suggesting that the single-reference-based coupled cluster theory is capable of providing reliable IE prediction for FeC, despite its multireference character. The CCSDTQ(Full)/CBS D0(Fe+−C) and D0(Fe−C) give the prediction of D0(Fe+−C) − D0(Fe−C) = 0.334 eV, which is consistent with the experimental determination of 0.3094 ± 0.0001 eV. The D0 calculations also support the experimental D0(Fe+−C) = 4.1 ± 0.3 eV and D0(Fe−C) = 3.8 ± 0.3 eV determined by the previous ion photodissociation study. The present calculations also provide the ΔHof0(ΔHof298) predictions for FeC/FeC+. The analysis of the correction terms in these calculations shows that the core−valence and valence−valence electronic correlations beyond CCSD(T) wave function and the relativistic effects make significant contributions to the calculated thermochemical properties of FeC/FeC+. For the experimental D0 and ΔHof0 values of FeC/FeC+, which are not known to high precision, we recommend the CCSDTQ(Full)/CBS predictions [D0(Fe−C) = 3.778 eV, D0(Fe+−C) = 4.112 eV, ΔHof0(FeC) = 760.8 kJ/mol and ΔHof0(FeC+) = 1490.6 kJ/mol] based on the ZPVE corrections using the experimental vibrational frequencies of FeC and FeC+.
Co-reporter:Shek-Man Yiu, Wai-Lun Man, Xin Wang, William W. Y. Lam, Siu-Mui Ng, Hoi-Ki Kwong, Kai-Chung Lau and Tai-Chu Lau
Chemical Communications 2011 - vol. 47(Issue 14) pp:NaN4161-4161
Publication Date(Web):2011/02/28
DOI:10.1039/C1CC00019E
MnO4− is activated by BF3 to undergo intramolecular coupling of two oxo ligands to generate O2. DFT calculations suggest that there should be a spin intercrossing between the singlet and triplet potential energy surfaces on going from the active intermediate [MnO2(OBF3)2]− to the O⋯O coupling transition state.
Co-reporter:Hoi-Ki Kwong, Po-Kam Lo, Kai-Chung Lau and Tai-Chu Lau
Chemical Communications 2011 - vol. 47(Issue 14) pp:NaN4275-4275
Publication Date(Web):2011/03/07
DOI:10.1039/C0CC05487A
The manganese(V) nitrido complex (PPh4)2[Mn(N)(CN)4] is an active catalyst for alkene epoxidation and alcohol oxidation using H2O2 as an oxidant. The catalytic oxidation is greatly enhanced by the addition of just one equivalent of acetic acid. The oxidation of ethene by this system has been studied computationally by the DFT method.
Co-reporter:Hongxia Du, Po-Kam Lo, Zongmin Hu, Haojun Liang, Kai-Chung Lau, Yi-Ning Wang, William W. Y. Lam and Tai-Chu Lau
Chemical Communications 2011 - vol. 47(Issue 25) pp:NaN7145-7145
Publication Date(Web):2011/05/25
DOI:10.1039/C1CC12024G
The oxidation of alcohols by KMnO4 is greatly accelerated by various Lewis acids. Notably the rate is increased by 4 orders of magnitude in the presence of Ca2+. The mechanisms of the oxidation of CH3OH and PhCH(OH)CH3 by MnO4− and BF3·MnO4− have also been studied computationally by the DFT method.
Co-reporter:Yih Chung Chang, Zhihong Luo, Yi Pan, Zheng Zhang, Ying-Nan Song, Sophie Yajin Kuang, Qing Zhu Yin, Kai-Chung Lau and C. Y. Ng
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 15) pp:NaN9793-9793
Publication Date(Web):2015/02/26
DOI:10.1039/C5CP00371G
By employing two-color visible (VIS)-ultraviolet (UV) laser photoionization and pulsed field ionization-photoelectron (PFI-PE) techniques, we have obtained highly rotationally resolved photoelectron spectra for vanadium monocarbide cations (VC+). The state-to-state VIS-UV-PFI-PE spectra thus obtained allow unambiguous assignments for the photoionization rotational transitions, resulting in a highly precise value for the adiabatic ionization energy (IE) of vanadium monocarbide (VC), IE(VC) = 57512.0 ± 0.8 cm−1 (7.13058 ± 0.00010 eV), which is defined as the energy of the VC+(X3Δ1; v+ = 0; J+ = 1) ← VC(X2Δ3/2; v′′ = 0; J′′ = 3/2) photoionization transition. The spectroscopic constants for VC+(X3Δ1) determined in the present study include the harmonic vibrational frequency ωe+ = 896.4 ± 0.8 cm−1, the anharmonicity constant ωe+xe+ = 5.7 ± 0.8 cm−1, the rotational constants Be+ = 0.6338 ± 0.0025 cm−1 and αe+ = 0.0033 ± 0.0007 cm−1, the equilibrium bond length re+ = 1.6549 ± 0.0003 Å, and the spin–orbit coupling constant A = 75.2 ± 0.8 cm−1 for VC+(X3Δ1,2,3). These highly precise energetic and spectroscopic data are used to benchmark state-of-the-art CCSDTQ/CBS calculations. In general, good agreement is found between the theoretical predictions and experimental results. The theoretical calculations yield the values, IE(VC) = 7.126 eV; the 0 K bond dissociation energies: D0(V–C) = 4.023 eV and D0(V+–C) = 3.663 eV; and heats of formation: , , , and kJ mol−1.

![1-Propyne, 3-[2,2,2-trifluoro-1,1-bis(trifluoromethyl)ethoxy]-](/data/chemimg/3723400/1632977-59-3.png)
![1-Propyne, 3-[2,2,2-trifluoro-1,1-bis(trifluoromethyl)ethoxy]-](/data/chemimg/3723400/1632977-59-3_b.png)

![Ethanone, 1-[3,5-bis(1,1-dimethylethyl)-2-methoxyphenyl]-](http://img.cochemist.com/ccimg/477800/477705-45-6.png)
![Ethanone, 1-[3,5-bis(1,1-dimethylethyl)-2-methoxyphenyl]-](http://img.cochemist.com/ccimg/477800/477705-45-6_b.png)
![Decacyclo[10.10.0.02,11.03,10.04,9.05,8.013,22.014,21.015,20.016,19]docosane](http://img.cochemist.com/ccimg/71700/71686-06-1.png)
![Decacyclo[10.10.0.02,11.03,10.04,9.05,8.013,22.014,21.015,20.016,19]docosane](http://img.cochemist.com/ccimg/71700/71686-06-1_b.png)




![tricyclo[4.2.0.02,5]octane](http://img.cochemist.com/ccimg/300/277-08-7.png)
![tricyclo[4.2.0.02,5]octane](http://img.cochemist.com/ccimg/300/277-08-7_b.png)




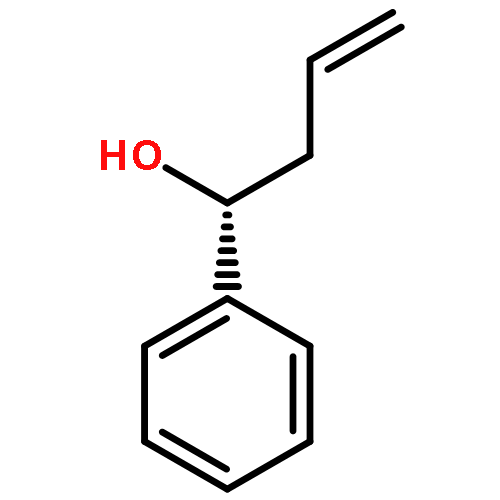
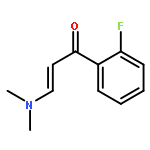
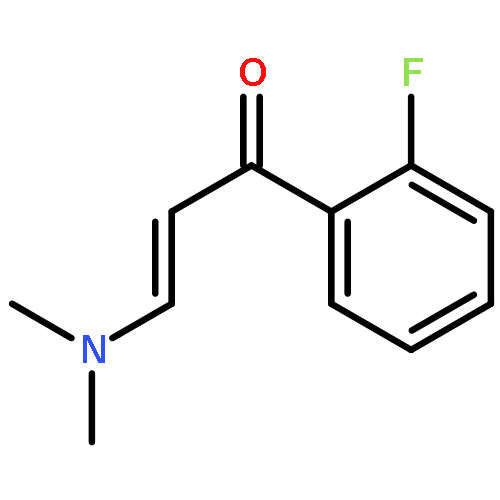





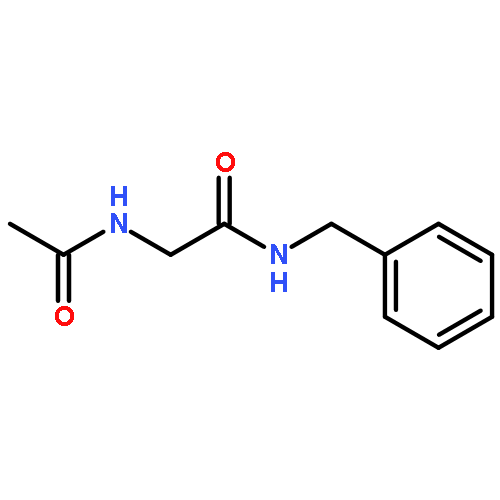








![2,2'-Bipyridine, 6-(5-[2,2'-bipyridin]-6-yl-1,1-dimethyl-3,4-diphenylsilacyclopenta-2,4-dien-2-yl)-](/data/chemimg/1202800/350042-00-1.png)
![2,2'-Bipyridine, 6-(5-[2,2'-bipyridin]-6-yl-1,1-dimethyl-3,4-diphenylsilacyclopenta-2,4-dien-2-yl)-](/data/chemimg/1202800/350042-00-1_b.png)









![Acetamide,N-[2-[[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/64200/64173-16-6.png)
![Acetamide,N-[2-[[[5-(dimethylamino)-1-naphthalenyl]sulfonyl]amino]ethyl]-](http://img.cochemist.com/ccimg/64200/64173-16-6_b.png)
![2,2'-Bipyridine, 6,6'-bis[(1R)-1-methoxy-2,2-dimethylpropyl]-](http://img.cochemist.com/ccimg/127100/127049-51-8.png)
![2,2'-Bipyridine, 6,6'-bis[(1R)-1-methoxy-2,2-dimethylpropyl]-](http://img.cochemist.com/ccimg/127100/127049-51-8_b.png)
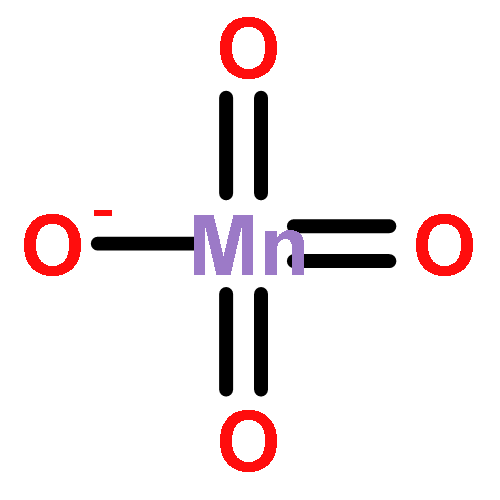
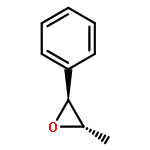
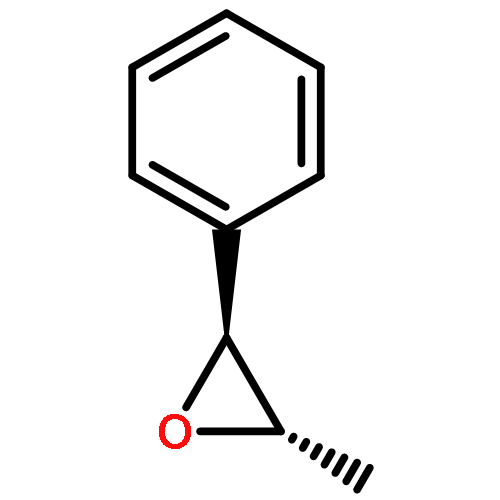


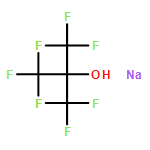
![Bicyclo[3.1.1]heptan-2-one, 6,6-dimethyl-3-methylene-, (1R,5R)-](http://img.cochemist.com/ccimg/34500/34413-89-3.png)
![Bicyclo[3.1.1]heptan-2-one, 6,6-dimethyl-3-methylene-, (1R,5R)-](http://img.cochemist.com/ccimg/34500/34413-89-3_b.png)












![3-(DIMETHYLAMINO)-1-[2-(TRIFLUOROMETHYL)PHENYL]PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/72900/72851-51-5.png)
![3-(DIMETHYLAMINO)-1-[2-(TRIFLUOROMETHYL)PHENYL]PROP-2-EN-1-ONE](http://img.cochemist.com/ccimg/72900/72851-51-5_b.png)





![{[(1R,2S,4R,6R)-1-METHYL-7-OXABICYCLO[4.1.0]HEPTANE-2,4-DIYL]BIS(<WBR />OXY)}BIS[DIMETHYL(2-METHYL-2-PROPANYL)SILANE]](http://img.cochemist.com/ccimg/324800/324753-11-9.png)
![{[(1R,2S,4R,6R)-1-METHYL-7-OXABICYCLO[4.1.0]HEPTANE-2,4-DIYL]BIS(<WBR />OXY)}BIS[DIMETHYL(2-METHYL-2-PROPANYL)SILANE]](http://img.cochemist.com/ccimg/324800/324753-11-9_b.png)














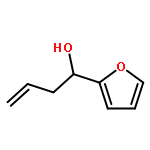
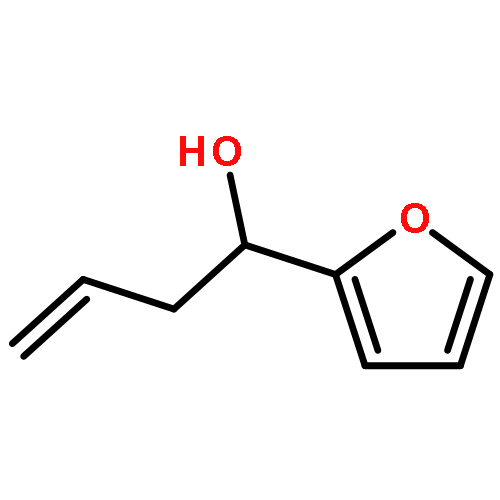




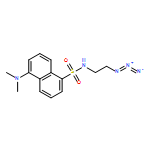
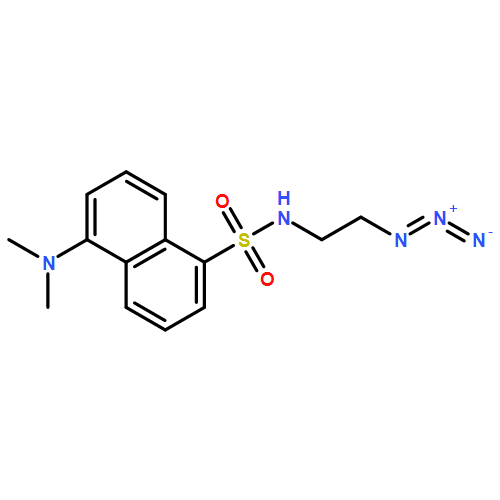






![Propanedinitrile, 2-[2-(1,1-dimethylethyl)-6-[2-(2,3,6,7-tetrahydro-1,1,7,7-tetramethyl-1H,5H-benzo[ij]quinolizin-9-yl)ethenyl]-4H-pyran-4-ylidene]-](/data/chemimg/2100/200052-70-6.png)
![Propanedinitrile, 2-[2-(1,1-dimethylethyl)-6-[2-(2,3,6,7-tetrahydro-1,1,7,7-tetramethyl-1H,5H-benzo[ij]quinolizin-9-yl)ethenyl]-4H-pyran-4-ylidene]-](/data/chemimg/2100/200052-70-6_b.png)



![2-Oxiraneoctanoic acid,3-[(3-pentyl-2-oxiranyl)methyl]-, methyl ester](http://img.cochemist.com/ccimg/2600/2500-56-3.png)
![2-Oxiraneoctanoic acid,3-[(3-pentyl-2-oxiranyl)methyl]-, methyl ester](http://img.cochemist.com/ccimg/2600/2500-56-3_b.png)

