Co-reporter:Bogdan Barnych;Amy A. Rand;Tomas Cajka;Kin Sing Stephen Lee
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 20) pp:4308-4313
Publication Date(Web):2017/05/23
DOI:10.1039/C7OB00789B
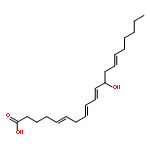
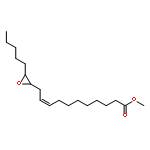
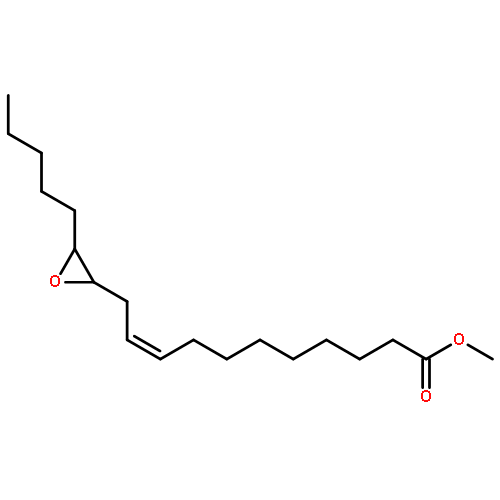
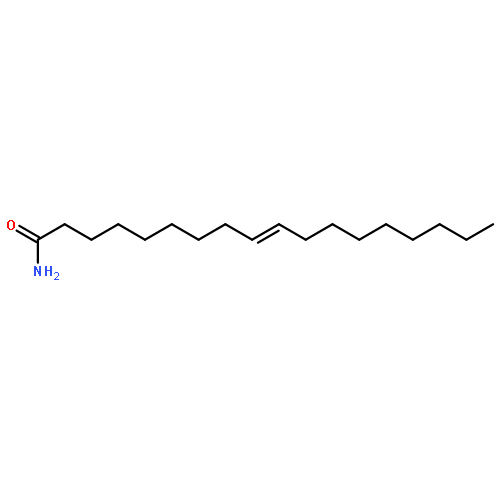
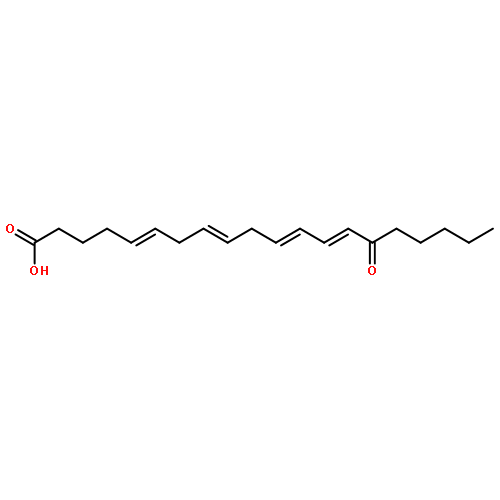
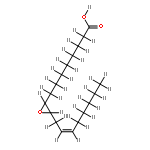
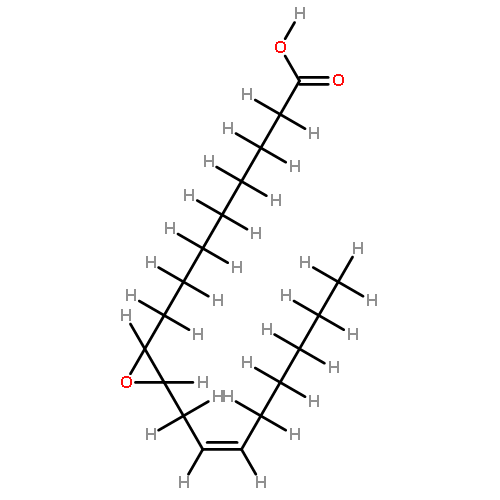
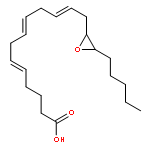
COX metabolites of 8,9-EET, previously observed as potent mitogenic lipid mediators, were synthesized for the first time by using two synthetic approaches. These synthetic materials allow for structural confirmation of COX metabolites of 8,9-EET and further study of their biological roles.
Co-reporter:Karen M. Wagner, Cindy B. McReynolds, William K. Schmidt, Bruce D. Hammock
Pharmacology & Therapeutics 2017 Volume 180(Volume 180) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.pharmthera.2017.06.006
Eicosanoids are biologically active lipid signaling molecules derived from polyunsaturated fatty acids. Many of the actions of eicosanoid metabolites formed by cyclooxygenase and lipoxygenase enzymes have been characterized, however, the epoxy-fatty acids (EpFAs) formed by cytochrome P450 enzymes are newly described by comparison. The EpFA metabolites modulate a diverse set of physiologic functions that include inflammation and nociception among others. Regulation of EpFAs occurs primarily via release, biosynthesis and enzymatic transformation by the soluble epoxide hydrolase (sEH). Targeting sEH with small molecule inhibitors has enabled observation of the biological activity of the EpFAs in vivo in animal models, greatly contributing to the overall understanding of their role in the inflammatory response. Their role in modulating inflammation has been demonstrated in disease models including cardiovascular pathology and inflammatory pain, but extends to neuroinflammation and neuroinflammatory disease. Moreover, while EpFAs demonstrate activity against inflammatory pain, interestingly, this action extends to blocking chronic neuropathic pain as well. This review outlines the role of modulating sEH and the biological action of EpFAs in models of pain and inflammatory diseases.
Co-reporter:Sumanta Kumar Goswami, Amelia Ann Rand, Debin Wan, Jun Yang, Bora Inceoglu, Melany Thomas, Christophe Morisseau, Guang-Yu Yang, Bruce D. Hammock
Life Sciences 2017 Volume 180(Volume 180) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.lfs.2017.05.018
AimsThis research was conducted to evaluate the hypothesis that gastric ulcers caused by the NSAID diclofenac sodium (DCF) can be prevented by the soluble epoxide hydrolase inhibitor TPPU.Main methodsMice were administered a single dose of 10, 30 or 100 mg/kg of DCF. Once an ulcerative dose of DCF was chosen, mice were pretreated with TPPU for 7 days at 0.1 mg/kg to evaluate anti-ulcer effects of the sEH inhibitor on anatomy, histopathology, pH, inflammatory markers and epithelial apoptosis of stomachs.Key findingsDiclofenac caused ulceration of the stomach at a dose of 100 mg/kg and a time post dose of 6 h. Ulcers generated under these conditions were associated with a significant increase in the levels of TNF-α and IL-6 in serum and increased apoptosis compared to control mice. Pretreatment with TPPU resulted in a decrease of ulceration in mice treated with DCF with a significant decrease in the level of apoptosis, TNF-α and IL-6 in the serum in comparison to diclofenac-treated mice. TPPU did not affect the pH of the stomach, whereas omeprazole elevated the pH of the stomach as expected. A similar anti-ulcer effect was observed in sEH gene knockout mice treated with DCF.SignificanceThe sEH inhibitor TPPU decreases the NSAID-induced stomach ulcers.
Co-reporter:Qian Ren;Jun Yang;Kenji Hashimoto;Karen M. Wagner;Chun Yang;Wei Yao;Mei Han;Chao Dong;Christophe Morisseau;Tamaki Ishima;Ji-chun Zhang;Min Ma
PNAS 2016 Volume 113 (Issue 13 ) pp:E1944-E1952
Publication Date(Web):2016-03-29
DOI:10.1073/pnas.1601532113
Depression is a severe and chronic psychiatric disease, affecting 350 million subjects worldwide. Although multiple antidepressants
have been used in the treatment of depressive symptoms, their beneficial effects are limited. The soluble epoxide hydrolase
(sEH) plays a key role in the inflammation that is involved in depression. Thus, we examined here the role of sEH in depression.
In both inflammation and social defeat stress models of depression, a potent sEH inhibitor, TPPU, displayed rapid antidepressant
effects. Expression of sEH protein in the brain from chronically stressed (susceptible) mice was higher than of control mice.
Furthermore, expression of sEH protein in postmortem brain samples of patients with psychiatric diseases, including depression,
bipolar disorder, and schizophrenia, was higher than controls. This finding suggests that increased sEH levels might be involved
in the pathogenesis of certain psychiatric diseases. In support of this hypothesis, pretreatment with TPPU prevented the onset
of depression-like behaviors after inflammation or repeated social defeat stress. Moreover, sEH KO mice did not show depression-like
behavior after repeated social defeat stress, suggesting stress resilience. The sEH KO mice showed increased brain-derived
neurotrophic factor (BDNF) and phosphorylation of its receptor TrkB in the prefrontal cortex, hippocampus, but not nucleus
accumbens, suggesting that increased BDNF-TrkB signaling in the prefrontal cortex and hippocampus confer stress resilience.
All of these findings suggest that sEH plays a key role in the pathophysiology of depression, and that epoxy fatty acids,
their mimics, as well as sEH inhibitors could be potential therapeutic or prophylactic drugs for depression.
Co-reporter:Natalia Vasylieva, Ki Chang Ahn, Bogdan Barnych, Shirley J. Gee, and Bruce D. Hammock
Environmental Science & Technology 2015 Volume 49(Issue 16) pp:10038-10047
Publication Date(Web):July 21, 2015
DOI:10.1021/acs.est.5b01005
Phenylpyrazole insecticides such as fipronil have been used as replacements for organophosphates. The wide application of fipronil raises concern about environmental contamination and risk for fish, birds, and other nontargeted beings as well as human health. A sensitive, competitive indirect heterologous enzyme-linked immunosorbent assay (ELISA) was developed. Antibodies with different specificities to fipronil and its metabolites were produced. Two ELISAs having IC50 values of 0.58 ± 0.06 and 2.6 ± 0.4 ng/mL were developed. Design of different haptens and coating antigens resulted in two assays with distinct cross-reactivity patterns for structurally related compounds: 96, 38, and 101% versus 39, 1.4, and 25% for fipronil-sulfide, fipronil-detrifluoromethylsulfonyl, and fipronil-desulfinyl, respectively. Performance of the immunoassays was demonstrated by a recovery study from spiked water and human serum and urine matrices, giving recovery values in the range of 85–111% for different concentrations. The assays demonstrated good correlation in fipronil recovery with conventional LC-MS/MS analysis. The generic assay 2265 has the sensitivity to measure fipronil and its analogs in serum at levels relevant for exposure monitoring. The assays were used to analyze human urine samples obtained from exposure studies and serum samples from rats treated with a fipronil-containing diet.
Co-reporter:Vladimir Burmistrov, Christophe Morisseau, Dmitry Danilov, Todd R. Harris, Igor Dalinger, Irina Vatsadze, Tatiana Shkineva, Gennady M. Butov, Bruce D. Hammock
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 23) pp:5514-5519
Publication Date(Web):1 December 2015
DOI:10.1016/j.bmcl.2015.10.066
Adamantyl ureas are good soluble epoxide hydrolase (sEH) inhibitors; however they have limited solubility and rapid metabolism, thus limiting their usefulness in some therapeutic indications. Herein, we test the hypothesis that nodal substitution on the adamantane will help solubilize and stabilize the compounds. A series of compounds containing adamantane derivatives and isoxazole functional groups were developed. Overall, the presence of methyl on the nodal positions of adamantane yields higher water solubility than previously reported urea-based sEH inhibitors while maintaining high inhibition potency. However, it did not improve microsomal stability.
Co-reporter:Bora Inceoglu;Kin Sing Stephen Lee;Ahmed Bettaieb;Carlos A. Trindade da Silva;Fawaz G. Haj
PNAS 2015 Volume 112 (Issue 29 ) pp:9082-9087
Publication Date(Web):2015-07-21
DOI:10.1073/pnas.1510137112
Despite intensive effort and resulting gains in understanding the mechanisms underlying neuropathic pain, limited success
in therapeutic approaches have been attained. A recently identified, nonchannel, nonneurotransmitter therapeutic target for
pain is the enzyme soluble epoxide hydrolase (sEH). The sEH degrades natural analgesic lipid mediators, epoxy fatty acids
(EpFAs), therefore its inhibition stabilizes these bioactive mediators. Here we demonstrate the effects of EpFAs on diabetes
induced neuropathic pain and define a previously unknown mechanism of pain, regulated by endoplasmic reticulum (ER) stress.
The activation of ER stress is first quantified in the peripheral nervous system of type I diabetic rats. We demonstrate that
both pain and markers of ER stress are reversed by a chemical chaperone. Next, we identify the EpFAs as upstream modulators
of ER stress pathways. Chemical inducers of ER stress invariably lead to pain behavior that is reversed by a chemical chaperone
and an inhibitor of sEH. The rapid occurrence of pain behavior with inducers, equally rapid reversal by blockers and natural
incidence of ER stress in diabetic peripheral nervous system (PNS) argue for a major role of the ER stress pathways in regulating
the excitability of the nociceptive system. Understanding the role of ER stress in generation and maintenance of pain opens
routes to exploit this system for therapeutic purposes.
Co-reporter:Cristina López-Vicario;Verónica García-Alonso;José Alcaraz-Quiles;Esther Titos;Vicente Arroyo;Sung H. Hwang;Joan Clària;Bibiana Rius;Aritz Lopategi
PNAS 2015 Volume 112 (Issue 2 ) pp:536-541
Publication Date(Web):2015-01-13
DOI:10.1073/pnas.1422590112
Soluble epoxide hydrolase (sEH) is an emerging therapeutic target in a number of diseases that have inflammation as a common
underlying cause. sEH limits tissue levels of cytochrome P450 (CYP) epoxides derived from omega-6 and omega-3 polyunsaturated
fatty acids (PUFA) by converting these antiinflammatory mediators into their less active diols. Here, we explored the metabolic
effects of a sEH inhibitor (t-TUCB) in fat-1 mice with transgenic expression of an omega-3 desaturase capable of enriching tissues with endogenous omega-3 PUFA. These
mice exhibited increased CYP1A1, CYP2E1, and CYP2U1 expression and abundant levels of the omega-3–derived epoxides 17,18-epoxyeicosatetraenoic
acid (17,18-EEQ) and 19,20-epoxydocosapentaenoic (19,20-EDP) in insulin-sensitive tissues, especially liver, as determined
by LC-ESI-MS/MS. In obese fat-1 mice, t-TUCB raised hepatic 17,18-EEQ and 19,20-EDP levels and reinforced the omega-3–dependent reduction observed in tissue inflammation
and lipid peroxidation. t-TUCB also produced a more intense antisteatotic action in obese fat-1 mice, as revealed by magnetic resonance spectroscopy. Notably, t-TUCB skewed macrophage polarization toward an antiinflammatory M2 phenotype and expanded the interscapular brown adipose
tissue volume. Moreover, t-TUCB restored hepatic levels of Atg12-Atg5 and LC3-II conjugates and reduced p62 expression, indicating up-regulation of
hepatic autophagy. t-TUCB consistently reduced endoplasmic reticulum stress demonstrated by the attenuation of IRE-1α and eIF2α phosphorylation.
These actions were recapitulated in vitro in palmitate-primed hepatocytes and adipocytes incubated with 19,20-EDP or 17,18-EEQ.
Relatively similar but less pronounced actions were observed with the omega-6 epoxide, 14,15-EET, and nonoxidized DHA. Together,
these findings identify omega-3 epoxides as important regulators of inflammation and autophagy in insulin-sensitive tissues
and postulate sEH as a druggable target in metabolic diseases.
Co-reporter:Aaron T. Wecksler;Sung Hee Hwang;Jun-Yan Liu;Hiromi I. Wettersten;Jian Wu;Robert H. Weiss;Christophe Morisseau;Bruce D. Hammock
Cancer Chemotherapy and Pharmacology 2015 Volume 75( Issue 1) pp:161-171
Publication Date(Web):2015/01/01
DOI:10.1007/s00280-014-2626-2
Sorafenib (Nexavar®) is currently the only FDA-approved small molecule targeted therapy for advanced hepatocellular carcinoma. The use of structural analogues and derivatives of sorafenib has enabled the elucidation of critical targets and mechanism(s) of cell death for human cancer lines. We previously performed a structure–activity relationship study on a series of sorafenib analogues designed to investigate the inhibition overlap between the major targets of sorafenib Raf-1 kinase and VEGFR-2, and an enzyme shown to be a potent off-target of sorafenib, soluble epoxide hydrolase. In the current work, we present the biological data on our lead sorafenib analogue, t-CUPM, demonstrating that this analogue retains cytotoxicity similar to sorafenib in various human cancer cell lines and strongly inhibits growth in the NCI-60 cell line panel. Co-treatment with the pan-caspase inhibitor, Z-VAD-FMK, failed to rescue the cell viability responses of both sorafenib and t-CUPM, and immunofluorescence microscopy shows similar mitochondrial depolarization and apoptosis-inducing factor release for both compounds. These data suggest that both compounds induce a similar mechanism of caspase-independent apoptosis in hepatoma cells. In addition, t-CUPM displays anti-proliferative effects comparable to sorafenib as seen by a halt in G0/G1 in cell cycle progression. The structural difference between sorafenib and t-CUPM significantly reduces inhibitory spectrum of kinases by this analogue, and pharmacokinetic characterization demonstrates a 20-fold better oral bioavailability of t-CUPM than sorafenib in mice. Thus, t-CUPM may have the potential to reduce the adverse events observed from the multikinase inhibitory properties and the large dosing regimens of sorafenib.
Co-reporter:Kin Sing Stephen Lee ; Jun-Yan Liu ; Karen M. Wagner ; Svetlana Pakhomova ; Hua Dong ; Christophe Morisseau ; Samuel H. Fu ; Jun Yang ; Peng Wang ; Arzu Ulu ; Christina A. Mate ; Long V. Nguyen ; Sung Hee Hwang ; Matthew L. Edin ; Alexandria A. Mara ; Heike Wulff ; Marcia E. Newcomer ; Darryl C. Zeldin
Journal of Medicinal Chemistry 2014 Volume 57(Issue 16) pp:7016-7030
Publication Date(Web):July 31, 2014
DOI:10.1021/jm500694p
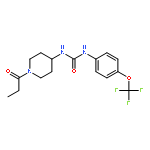
Diabetes is affecting the life of millions of people. A large proportion of diabetic patients suffer from severe complications such as neuropathic pain, and current treatments for these complications have deleterious side effects. Thus, alternate therapeutic strategies are needed. Recently, the elevation of epoxy-fatty acids through inhibition of soluble epoxide hydrolase (sEH) was shown to reduce diabetic neuropathic pain in rodents. In this report, we describe a series of newly synthesized sEH inhibitors with at least 5-fold higher potency and doubled residence time inside both the human and rodent sEH enzyme than previously reported inhibitors. These inhibitors also have better physical properties and optimized pharmacokinetic profiles. The optimized inhibitor selected from this new series displayed improved efficacy of almost 10-fold in relieving pain perception in diabetic neuropathic rats as compared to the approved drug, gabapentin, and previously published sEH inhibitors. Therefore, these new sEH inhibitors could be an attractive alternative to treat diabetic neuropathy in humans.
Co-reporter:Candace R. S. Bever, Zuzana Majkova, Rajeswaran Radhakrishnan, Ian Suni, Mark McCoy, Yanru Wang, Julie Dechant, Shirley Gee, and Bruce D. Hammock
Analytical Chemistry 2014 Volume 86(Issue 15) pp:7875
Publication Date(Web):June 23, 2014
DOI:10.1021/ac501807j
An antibody-based analytical method for the detection of a chemical flame retardant using antibody fragments isolated from an alpaca has been developed. One specific chemical flame retardant congener, 2,2′,4,4′-tetrabrominated diphenyl ether (BDE-47), is often the major poly-BDE (PBDE) congener present in human and environmental samples and that which is the most frequently detected. An alpaca was immunized with a surrogate of BDE-47 covalently attached to a carrier protein. The resulting mRNA coding for the variable domain of heavy-chain antibodies (VHH) were isolated, transcribed to cDNA, and cloned into a phagemid vector for phage display library construction. Selection of VHHs recognizing BDE-47 was achieved by panning under carefully modified conditions. The assay sensitivity for detecting BDE-47 was down to the part-per-billion (microgram per liter) level. Cross-reactivity analyses confirmed that this method was highly selective for BDE-47 and selected hydroxylated metabolites. When exposed to elevated temperatures, the camelid VHH antibodies retained more reactivity than a polyclonal antibody developed to the same target analyte. The use of this VHH antibody reagent immobilized onto a Au electrode for impedance biosensing demonstrates the increased versatility of VHH antibodies.
Co-reporter:Vladimir Burmistrov, Christophe Morisseau, Kin Sing Stephen Lee, Diyala S. Shihadih, Todd R. Harris, Gennady M. Butov, Bruce D. Hammock
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 9) pp:2193-2197
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmcl.2014.03.016
A series of inhibitors of the soluble epoxide hydrolase (sEH) containing two urea groups has been developed. Inhibition potency of the described compounds ranges from 2.0 μM to 0.4 nM. 1,6-(Hexamethylene)bis[(adamant-1-yl)urea] (3b) was found to be a potent slow tight binding inhibitor (IC50 = 0.5 nM) with a strong binding to sEH (Ki = 3.1 nM) and a moderately long residence time on the enzyme (koff = 1.05 × 10−3 s−1; t1/2 = 11 min).
Co-reporter:Koji Taniguchi;Ronald M. Evans;Mei-Fei Yueh;Shujuan Chen;Michael Karin;Robert H. Tukey
PNAS 2014 Volume 111 (Issue 48 ) pp:17200-17205
Publication Date(Web):2014-12-02
DOI:10.1073/pnas.1419119111
Triclosan [5-chloro-2-(2,4-dichlorophenoxy)phenol; TCS] is a synthetic, broad-spectrum antibacterial chemical used in a wide
range of consumer products including soaps, cosmetics, therapeutics, and plastics. The general population is exposed to TCS
because of its prevalence in a variety of daily care products as well as through waterborne contamination. TCS is linked to
a multitude of health and environmental effects, ranging from endocrine disruption and impaired muscle contraction to effects
on aquatic ecosystems. We discovered that TCS was capable of stimulating liver cell proliferation and fibrotic responses,
accompanied by signs of oxidative stress. Through a reporter screening assay with an array of nuclear xenobiotic receptors
(XenoRs), we found that TCS activates the nuclear receptor constitutive androstane receptor (CAR) and, contrary to previous
reports, has no significant effect on mouse peroxisome proliferation activating receptor α (PPARα). Using the procarcinogen
diethylnitrosamine (DEN) to initiate tumorigenesis in mice, we discovered that TCS substantially accelerates hepatocellular
carcinoma (HCC) development, acting as a liver tumor promoter. TCS-treated mice exhibited a large increase in tumor multiplicity,
size, and incidence compared with control mice. TCS-mediated liver regeneration and fibrosis preceded HCC development and
may constitute the primary tumor-promoting mechanism through which TCS acts. These findings strongly suggest there are adverse
health effects in mice with long-term TCS exposure, especially on enhancing liver fibrogenesis and tumorigenesis, and the
relevance of TCS liver toxicity to humans should be evaluated.
Co-reporter:Chunqing Zhao;Sung Hee Hwang;Bruce A. Buchholz;Timothy S. Carpenter;Felice C. Lightstone;Jun Yang;John E. Casida
PNAS 2014 111 (23 ) pp:8607-8612
Publication Date(Web):2014-06-10
DOI:10.1073/pnas.1407379111
Use of the highly toxic and easily prepared rodenticide tetramethylenedisulfotetramine (TETS) was banned after thousands of
accidental or intentional human poisonings, but it is of continued concern as a chemical threat agent. TETS is a noncompetitive
blocker of the GABA type A receptor (GABAAR), but its molecular interaction has not been directly established for lack of a suitable radioligand to localize the binding
site. We synthesized [14C]TETS (14 mCi/mmol, radiochemical purity >99%) by reacting sulfamide with H14CHO and s-trioxane then completion of the sequential cyclization with excess HCHO. The outstanding radiocarbon sensitivity of accelerator
mass spectrometry (AMS) allowed the use of [14C]TETS in neuroreceptor binding studies with rat brain membranes in comparison with the standard GABAAR radioligand 4′-ethynyl-4-n-[3H]propylbicycloorthobenzoate ([3H]EBOB) (46 Ci/mmol), illustrating the use of AMS for characterizing the binding sites of high-affinity 14C radioligands. Fourteen noncompetitive antagonists of widely diverse chemotypes assayed at 1 or 10 µM inhibited [14C]TETS and [3H]EBOB binding to a similar extent (r2 = 0.71). Molecular dynamics simulations of these 14 toxicants in the pore region of the α1β2γ2 GABAAR predict unique and significant polar interactions for TETS with α1T1′ and γ2S2′, which are not observed for EBOB or the GABAergic insecticides. Several GABAAR modulators similarly inhibited [14C]TETS and [3H]EBOB binding, including midazolam, flurazepam, avermectin Ba1, baclofen, isoguvacine, and propofol, at 1 or 10 μM, providing
an in vitro system for recognizing candidate antidotes.
Co-reporter:Guodong Zhang;Dipak Panigrahy;Sung Hee Hwang;Lisa M. Mahakian;Jun Yang;Hiromi I. Wettersten;Jun-Yan Liu;Yanru Wang;Elizabeth S. Ingham;Sarah Tam;Mark W. Kieran;Robert H. Weiss;Katherine W. Ferrara
PNAS 2014 Volume 111 (Issue 30 ) pp:11127-11132
Publication Date(Web):2014-07-29
DOI:10.1073/pnas.1410432111
Prostaglandins derived from the cyclooxygenase (COX) pathway and epoxyeicosatrienoic acids (EETs) from the cytochrome P450/soluble
epoxide hydrolase (sEH) pathway are important eicosanoids that regulate angiogenesis and tumorigenesis. COX-2 inhibitors,
which block the formation of prostaglandins, suppress tumor growth, whereas sEH inhibitors, which increase endogenous EETs,
stimulate primary tumor growth and metastasis. However, the functional interactions of these two pathways in cancer are unknown.
Using pharmacological inhibitors as probes, we show here that dual inhibition of COX-2 and sEH synergistically inhibits primary
tumor growth and metastasis by suppressing tumor angiogenesis. COX-2/sEH dual pharmacological inhibitors also potently suppress
primary tumor growth and metastasis by inhibiting tumor angiogenesis via selective inhibition of endothelial cell proliferation.
These results demonstrate a critical interaction of these two lipid metabolism pathways on tumorigenesis and suggest dual
inhibition of COX-2 and sEH as a potential therapeutic strategy for cancer therapy.
Co-reporter:Anupama Ranganathan, Grace A. Paradise, Chad A. Hansen, Mark R. McCoy, Shirley J. Gee, Ping Zhong, Dan Chang, and Bruce D. Hammock
Journal of Agricultural and Food Chemistry 2013 Volume 61(Issue 28) pp:6964-6970
Publication Date(Web):June 14, 2013
DOI:10.1021/jf401302y
Hesperetin dihydrochalcone 4′-glucoside, 1, and phloretin 4′-glucoside, 2, belong to a family of dihydrochalcone glycosides that exhibit flavorant properties. In this study was developed a competitive, indirect homologous ELISA for the detection of targets 1 and 2 in fermentation media. Immunogen and coating antigen were prepared by conjugating hapten, 4-(3-oxo-3-(2,6-dihydroxy-4-glucoside phenyl)propyl) benzoic acid, to thyroglobulin and bovine serum albumin, respectively. Antibodies raised in rabbits M6122, M6123, and M6124 and the coating antigen were screened and characterized to determine their optimum concentrations. The optimized ELISA, developed with antibody M6122, gave IC50 values of 27.8 and 21.8 ng/mL for 1 and 2, respectively. Selectivity of the assay was assessed by measuring cross-reactivity of antibody M6122 to related congeners such as aglycones and the 2′-glycosides of hesperetin dihydrochalcone, 5 and phloretin, 6. Antibody M6122 showed very low recognition of 5 and virtually no recognition of the aglycones and 6.
Co-reporter:Kin Sing Stephen Lee, Christophe Morisseau, Jun Yang, Peng Wang, Sung Hee Hwang, Bruce D. Hammock
Analytical Biochemistry 2013 Volume 434(Issue 2) pp:259-268
Publication Date(Web):15 March 2013
DOI:10.1016/j.ab.2012.11.015
The soluble epoxide hydrolase (sEH), responsible for the hydrolysis of various fatty acid epoxides to their corresponding 1,2-diols, is becoming an attractive pharmaceutical target. These fatty acid epoxides, particularly epoxyeicosatrienoic acids (EETs), play an important role in human homeostatic and inflammation processes. Therefore, inhibition of human sEH, which stabilizes EETs in vivo, brings several beneficial effects to human health. Although there are several catalytic assays available to determine the potency of sEH inhibitors, measuring the in vitro inhibition constant (Ki) for these inhibitors using catalytic assay is laborious. In addition, koff, which has been recently suggested to correlate better with the in vivo potency of inhibitors, has never been measured for sEH inhibitors. To better measure the potency of sEH inhibitors, a reporting ligand, 1-(adamantan-1-yl)-3-(1-(2-(7-hydroxy-2-oxo-2H-chromen-4-yl)acetyl) piperidin-4-yl)urea (ACPU), was designed and synthesized. With ACPU, we have developed a Förster resonance energy transfer (FRET)-based competitive displacement assay using intrinsic tryptophan fluorescence from sEH. In addition, the resulting assay allows us to measure the Ki values of very potent compounds to the picomolar level and to obtain relative koff values of the inhibitors. This assay provides additional data to evaluate the potency of sEH inhibitors.
Co-reporter:Sung Hee Hwang, Aaron T. Wecksler, Guodong Zhang, Christophe Morisseau, Long V. Nguyen, Samuel H. Fu, Bruce D. Hammock
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 13) pp:3732-3737
Publication Date(Web):1 July 2013
DOI:10.1016/j.bmcl.2013.05.011
To reduce the pro-angiogenic effects of sEH inhibition, a structure–activity relationship (SAR) study was performed by incorporating structural features of the anti-angiogenic multi-kinase inhibitor sorafenib into soluble epoxide hydrolase (sEH) inhibitors. The structural modifications of this series of molecules enabled the altering of selectivity towards the pro-angiogenic kinases C-RAF and vascular endothelial growth factor receptor-2 (VEGFR-2), while retaining their sEH inhibition. As a result, sEH inhibitors with greater potency against C-RAF and VEGFR-2 were obtained. Compound 4 (t-CUPM) possesses inhibition potency higher than sorafenib towards sEH but similar against C-RAF and VEGFR-2. Compound 7 (t-CUCB) selectively inhibits sEH, while inhibiting HUVEC cell proliferation, a potential anti-angiogenic property, without liver cancer cell cytotoxicity. The data presented suggest a potential rational approach to control the angiogenic responses stemming from sEH inhibition.
Co-reporter:Shizuo G. Kamita;Grant H. Oshita;Peng Wang;Christophe Morisseau;Raja Sekhar Nety;Bryce W. Falk
Archives of Insect Biochemistry and Physiology 2013 Volume 83( Issue 4) pp:171-179
Publication Date(Web):
DOI:10.1002/arch.21100
Epoxide hydrolase (EH) is an enzyme in the α/β-hydrolase fold superfamily that uses a water molecule to transform an epoxide to its corresponding diol. In insects, EHs metabolize among other things critical developmental hormones called juvenile hormones (JHs). EHs also play roles in the detoxification of toxic compounds that are found in the insect's diet or environment. In this study, a full-length cDNA encoding an epoxide hydrolase, Hovi-mEH1, was obtained from the xylem-feeding insect Homalodisca vitripennis. H. vitripennis, commonly known as the glassy-winged sharpshooter, is an economically important vector of plant pathogenic bacteria such as Xylella fastidiosa. Hovi-mEH1 hydrolyzed the general EH substrates cis-stilbene oxide and trans-diphenylpropene oxide with specific activities of 47.5 ± 6.2 and 1.3 ± 0.5 nmol of diol formed min−1 mg−1, respectively. Hovi-mEH1 metabolized JH III with a Vmax of 29.3 ± 1.6 nmol min−1 mg−1, kcat of 0.03 s−1, and KM of 13.8 ± 2.0 μM. These Vmax and kcat values are similar to those of known JH metabolizing EHs from lepidopteran and coleopteran insects. Hovi-mEH1 showed 99.1% identity to one of three predicted EH-encoding sequences that were identified in the transcriptome of H. vitripennis. Of these three sequences only Hovi-mEH1 clustered with known JH metabolizing EHs. On the basis of biochemical, phylogenetic, and transcriptome analyses, we hypothesize that Hovi-mEH1 is a biologically relevantJH-metabolizing enzyme in H. vitripennis.
Co-reporter:Brian T. Kalish;Dipak Panigrahy;Sui Huang;Diane R. Bielenberg;Hau D. Le;Jun Yang;Matthew L. Edin;Dayna K. Mudge;Akiko Mammoto;Ofra Benny;Roger L. Jenkins;Mary A. Simpson;Catherine E. Butterfield;Bora Inceoglu;Craig R. Lee;Tomoshige Akino;Tadanori Mammoto;Kenneth B. Tomer;Fred B. Lih;Donald E. Ingber;John R. Falck;Vijaya L. Manthati;Arja Kaipainen;Patricia A. D’Amore;Mark Puder;Darryl C. Zeldin;Mark W. Kieran
PNAS 2013 Volume 110 (Issue 33 ) pp:13528-13533
Publication Date(Web):2013-08-13
DOI:10.1073/pnas.1311565110
Epoxyeicosatrienoic acids (EETs), lipid mediators produced by cytochrome P450 epoxygenases, regulate inflammation, angiogenesis,
and vascular tone. Despite pleiotropic effects on cells, the role of these epoxyeicosanoids in normal organ and tissue regeneration
remains unknown. EETs are produced predominantly in the endothelium. Normal organ and tissue regeneration require an active
paracrine role of the microvascular endothelium, which in turn depends on angiogenic growth factors. Thus, we hypothesize
that endothelial cells stimulate organ and tissue regeneration via production of bioactive EETs. To determine whether endothelial-derived
EETs affect physiologic tissue growth in vivo, we used genetic and pharmacological tools to manipulate endogenous EET levels.
We show that endothelial-derived EETs play a critical role in accelerating tissue growth in vivo, including liver regeneration,
kidney compensatory growth, lung compensatory growth, wound healing, corneal neovascularization, and retinal vascularization.
Administration of synthetic EETs recapitulated these results, whereas lowering EET levels, either genetically or pharmacologically,
delayed tissue regeneration, demonstrating that pharmacological modulation of EETs can affect normal organ and tissue growth.
We also show that soluble epoxide hydrolase inhibitors, which elevate endogenous EET levels, promote liver and lung regeneration.
Thus, our observations indicate a central role for EETs in organ and tissue regeneration and their contribution to tissue
homeostasis.
Co-reporter:Guodong Zhang;Jun-Yan Liu;Dipak Panigrahy;Jun Yang;Lisa M. Mahakian;Arzu Ulu;Xiaowen Hu;Elizabeth S. Ingham;Mark W. Kieran;Kin Sing Stephen Lee;Sung Hee Hwang;Robert H. Weiss;Hiromi I. Wettersten;Sarah Tam;Katherine W. Ferrara
PNAS 2013 Volume 110 (Issue 16 ) pp:6530-6535
Publication Date(Web):2013-04-16
DOI:10.1073/pnas.1304321110
Epidemiological and preclinical evidence supports that omega-3 dietary fatty acids (fish oil) reduce the risks of macular
degeneration and cancers, but the mechanisms by which these omega-3 lipids inhibit angiogenesis and tumorigenesis are poorly
understood. Here we show that epoxydocosapentaenoic acids (EDPs), which are lipid mediators produced by cytochrome P450 epoxygenases
from omega-3 fatty acid docosahexaenoic acid, inhibit VEGF- and fibroblast growth factor 2-induced angiogenesis in vivo, and
suppress endothelial cell migration and protease production in vitro via a VEGF receptor 2-dependent mechanism. When EDPs
(0.05 mg⋅kg−1⋅d−1) are coadministered with a low-dose soluble epoxide hydrolase inhibitor, EDPs are stabilized in circulation, causing ∼70%
inhibition of primary tumor growth and metastasis. Contrary to the effects of EDPs, the corresponding metabolites derived
from omega-6 arachidonic acid, epoxyeicosatrienoic acids, increase angiogenesis and tumor progression. These results designate
epoxyeicosatrienoic acids and EDPs as unique endogenous mediators of an angiogenic switch to regulate tumorigenesis and implicate
a unique mechanistic linkage between omega-3 and omega-6 fatty acids and cancers.
Co-reporter:Hong C. Shen
Journal of Medicinal Chemistry 2012 Volume 55(Issue 5) pp:1789-1808
Publication Date(Web):December 14, 2011
DOI:10.1021/jm201468j
Co-reporter:Hee-Joo Kim, Mark R. McCoy, Zuzana Majkova, Julie E. Dechant, Shirley J. Gee, Sofia Tabares-da Rosa, Gualberto G. González-Sapienza, and Bruce D. Hammock
Analytical Chemistry 2012 Volume 84(Issue 2) pp:1165
Publication Date(Web):December 12, 2011
DOI:10.1021/ac2030255
Some unique subclasses of Camelidae antibodies are devoid of the light chain, and the antigen binding site is comprised exclusively of the variable domain of the heavy chain (VHH). Although conventional antibodies dominate current assay development, recombinant VHHs have a high potential as alternative reagents for the next generation of immunoassay. We expressed VHHs from an immunized alpaca and developed a VHH-based immunoassay using 3-phenoxybenzoic acid (3-PBA), a major metabolite of pyrethroid insecticides as a model system. A phage VHH library was constructed, and seven VHH clones were selected by competitive binding with 3-PBA. The best immunoassay developed with one of these VHHs showed an IC50 of 1.4 ng/mL (limit of detection (LOD) = 0.1 ng/mL). These parameters were further improved by using the phage borne VHH, IC50 = 0.1 ng/mL and LOD = 0.01 ng/mL. Both assays showed a similar tolerance to methanol and dimethylsulfoxide up to 50% in assay buffer. The assay was highly specific to 3-PBA and its 4-hydroxylated derivative, 4-hydroxy 3-PBA, (150% cross reactivity) with negligible cross reactivity with other tested structural analogues, and the recovery from spiked urine sample ranged from 80 to 112%. In conclusion, a highly specific and sensitive VHH for 3-PBA was developed using sequences from immunized alpaca and phage display technology for antibody selection.
Co-reporter:A Ulu;SE Appt;C Morisseau;SH Hwang;PD Jones;TE Rose;H Dong;J Lango;J Yang;HJ Tsai;C Miyabe;C Fortenbach;MR Adams;BD Hammock
British Journal of Pharmacology 2012 Volume 165( Issue 5) pp:1401-1412
Publication Date(Web):
DOI:10.1111/j.1476-5381.2011.01641.x
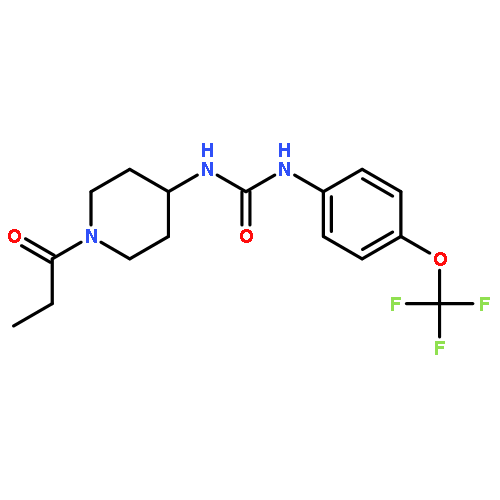
BACKGROUND AND PURPOSE Soluble epoxide hydrolase inhibitors (sEHIs) possess anti-inflammatory, antiatherosclerotic, antihypertensive and analgesic properties. The pharmacokinetics (PK) and pharmacodynamics in terms of inhibitory potency of sEHIs were assessed in non-human primates (NHPs). Development of a sEHI for use in NHPs will facilitate investigations on the role of sEH in numerous chronic inflammatory conditions.
EXPERIMENTAL APPROACH PK parameters of 11 sEHIs in cynomolgus monkeys were determined after oral dosing with 0.3 mg·kg−1. Their physical properties and inhibitory potency in hepatic cytosol of cynomolgus monkeys were examined. Dose-dependent effects of the two inhibitors 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) and the related acetyl piperidine derivative, 1-trifluoromethoxyphenyl-3-(1-acetylpiperidin-4-yl) urea (TPAU), on natural blood eicosanoids, were determined.
KEY RESULTS Among the inhibitors tested, TPPU and two 4-(cyclohexyloxy) benzoic acid urea sEHIs displayed high plasma concentrations (>10 × IC50), when dosed orally at 0.3 mg·kg−1. Although the 4-(cyclohexyloxy) benzoic acid ureas were more potent against monkey sEH than piperidyl ureas (TPAU and TPPU), the latter compounds showed higher plasma concentrations and more drug-like properties. The Cmax increased with dose from 0.3 to 3 mg·kg−1 for TPPU and from 0.1 to 3 mg·kg−1 for TPAU, although it was not linear over this range of doses. As an indication of target engagement, ratios of linoleate epoxides to diols increased with TPPU administration.
CONCLUSION AND IMPLICATIONS Our data indicate that TPPU is suitable for investigating sEH biology and the role of epoxide-containing lipids in modulating inflammatory diseases in NHPs.
Co-reporter:Ki Chang Ahn, Takeo Kasagami, Hsing-Ju Tsai, Nils Helge Schebb, Temitope Ogunyoku, Shirley J. Gee, Thomas M. Young, and Bruce D. Hammock
Environmental Science & Technology 2012 Volume 46(Issue 1) pp:374-381
Publication Date(Web):November 11, 2011
DOI:10.1021/es202494d
A sensitive, competitive indirect enzyme-linked immunosorbent assay (ELISA) for the detection of the antimicrobial triclocarban (TCC) was developed. The haptens were synthesized by derivatizing the para position of a phenyl moiety of TCC. The rabbit antisera were screened and the combination of antiserum 1648 and a heterologous competitive hapten containing a piperidine was further characterized. The IC50 and detection range for TCC in buffer were 0.70 and 0.13–3.60 ng/mL, respectively. The assay was selective for TCC, providing only low cross-reactivity to TCC-related compounds and its major metabolites except for the closely related antimicrobial 3-trifluoromethyl-4,4′-dichlorocarbanilide. A liquid–liquid extraction for sample preparation of human body fluids resulted in an assay that measured low part per billion levels of TCC in small volumes of the samples. The limits of quantification of TCC were 5 ng/mL in blood/serum and 10 ng/mL in urine, respectively. TCC in human urine was largely the N- or N′-glucuronide. TCC concentrations of biosolids measured by the ELISA were similar to those determined by LC-MS/MS. This immunoassay can be used as a rapid, inexpensive, and convenient tool to aid researchers monitoring human/environmental exposure to TCC to better understand the health effects.
Co-reporter:Mark R. McCoy, Zheng Yang, Xun Fu, Ki Chang Ahn, Shirley J. Gee, David C. Bom, Ping Zhong, Dan Chang, and Bruce D. Hammock
Journal of Agricultural and Food Chemistry 2012 Volume 60(Issue 20) pp:5065-5070
Publication Date(Web):April 9, 2012
DOI:10.1021/jf2051653
Pyrethroids are a class of insecticides that are becoming increasingly popular in agricultural and home use applications. Sensitive assays for pyrethroid insecticides in complex matrices are difficult with both instrumental and immunochemical methods. Environmental analysis of the pyrethroids by immunoassay requires either knowing which pyrethroids contaminate the source or the use of nonspecific antibodies with cross-reactivities to a class of compounds. We describe an alternative method that converts the type II pyrethroids to a common chemical product, 3-phenoxybenzoic acid (3-PBA), prior to analysis. This method is much more sensitive than detecting the parent compound, and it is much easier to detect a single compound rather than an entire class of compounds. This is useful in screening for pyrethroids as a class or in situations where a single type of pyrethroid is used. We demonstrated this technique in both citrus oils and environmental water samples with conversion rates of the pyrethroid to 3-PBA that range from 45 to 75% and methods that require no extraction steps for either the immunoassay or the liquid chromatography–tandem mass spectrometry (LC-MS/MS) techniques. Limits of detection for this technique applied to orange oil are 5 nM, 2 μM, and 0.8 μM when detected by LC-MS/MS, gas chromatography–mass spectrometry, and immunoassay, respectively. The limit of detection for pyrethroids in water when detected by immunoassay was 2 nM.
Co-reporter:In-Hae Kim, Kosuke Nishi, Takeo Kasagami, Christophe Morisseau, Jun-Yan Liu, Hsing-Ju Tsai, Bruce D. Hammock
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 18) pp:5889-5892
Publication Date(Web):15 September 2012
DOI:10.1016/j.bmcl.2012.07.074
Substituted ureas with a carboxylic acid ester as a secondary pharmacophore are potent soluble epoxide hydrolase (sEH) inhibitors. Although the ester substituent imparts better physical properties, such compounds are quickly metabolized to the corresponding less potent acids. Toward producing biologically active ester compounds, a series of esters were prepared and evaluated for potency on the human enzyme, stability in human liver microsomes, and physical properties. Modifications around the ester function enhanced in vitro metabolic stability of the ester inhibitors up to 32-fold without a decrease in inhibition potency. Further, several compounds had improved physical properties.
Co-reporter:Sarunya Thiphom, Tippawan Prapamontol, Somporn Chantara, Ampica Mangklabruks, Chaisuree Suphavilai, Ki Chang Ahn, Shirley J. Gee and Bruce D. Hammock
Analytical Methods 2012 vol. 4(Issue 11) pp:3772-3778
Publication Date(Web):03 Sep 2012
DOI:10.1039/C2AY25642H
The aim of this study was to identify a plasma biomarker of exposure to pyrethroid insecticides. A major metabolite, 3-phenoxybenzoic acid (3-PBA), can be detected in urine but urinary 3-PBA cannot be used to assess the active dose. The 3-PBA-adduct represents a much more persistent class of biomarkers than metabolites excreted into urine, having half-lives of up to several weeks or months. We developed an enzyme-linked immunosorbent assay (ELISA) for total 3-PBA including the adduct formed after alkaline hydrolysis, liquid–liquid extraction (LLE) and solid phase extraction (SPE) of the sample. The developed ELISA had an IC50 value of 26.7 ng mL−1. The intra- and inter-assay coefficients of variation (%CV) were lower than 5% and were within the optimum condition variance (OCV) range. The LLE cleanup technique satisfactorily eliminated the matrix effect from plasma samples before SPE and ELISA analysis yielding good recoveries (85.9–99.4%) with a limit of quantitation (LOQ, 5 ng mL−1) that was 30- to 47-fold more sensitive than previous studies. Moreover, the method developed could separate more than 80% of 3-PBA from the adducted form. The method was successfully applied for the detection of the target in real samples obtained from consumers (n = 50) and farmers (n = 50). To our knowledge, this is the first ELISA method for detecting 3-PBA in the human plasma that has been applied to a field study.
Co-reporter:Huazhang Huang, Hongwei Yao, Jun-Yan Liu, Aman I. Samra, Shizuo G. Kamita, Anthony J. Cornel, Bruce D. Hammock
Analytical Biochemistry 2012 Volume 431(Issue 2) pp:77-83
Publication Date(Web):15 December 2012
DOI:10.1016/j.ab.2012.09.011
The availability of highly sensitive substrates is critical for the development of precise and rapid assays for detecting changes in glutathione S-transferase (GST) activity that are associated with GST-mediated metabolism of insecticides. In this study, six pyrethroid-like compounds were synthesized and characterized as substrates for insect and mammalian GSTs. All of the substrates were esters composed of the same alcohol moiety, 7-hydroxy-4-methylcoumarin, and acid moieties that structurally mimic some commonly used pyrethroid insecticides, including cypermethrin and cyhalothrin. CpGSTD1, a recombinant Delta class GST from the mosquito Culex pipiens pipiens, metabolized our pyrethroid-like substrates with both chemical and geometric preference (i.e., the cis-isomers were metabolized at 2- to 5-fold higher rates than the corresponding trans-isomers). A GST preparation from mouse liver also metabolized most of our pyrethroid-like substrates with both chemical and geometric preference but at 10- to 170-fold lower rates. CpGSTD1 and mouse GSTs metabolized 1-chloro-2,4-dinitrobenezene (CDNB), a general GST substrate, at more than 200-fold higher rates than our novel pyrethroid-like substrates. There was a 10-fold difference in the specificity constant (kcat/KM ratio) of CpGSTD1 for CDNB and those of CpGSTD1 for cis-DCVC and cis-TFMCVC, suggesting that cis-DCVC and cis-TFMCVC may be useful for the detection of GST-based metabolism of pyrethroids in mosquitoes.
Co-reporter:Gennady Cherednichenko;Rui Zhang;Roger A. Bannister;Valeriy Timofeyev;Ning Li;Erika B. Fritsch;Wei Feng;Genaro C. Barrientos;Nils H. Schebb;Kurt G. Beam;Nipavan Chiamvimonvat;Isaac N. Pessah
PNAS 2012 109 (35 ) pp:14158-14163
Publication Date(Web):2012-08-28
DOI:10.1073/pnas.1211314109
Triclosan (TCS), a high-production-volume chemical used as a bactericide in personal care products, is a priority pollutant
of growing concern to human and environmental health. TCS is capable of altering the activity of type 1 ryanodine receptor
(RyR1), but its potential to influence physiological excitation–contraction coupling (ECC) and muscle function has not been
investigated. Here, we report that TCS impairs ECC of both cardiac and skeletal muscle in vitro and in vivo. TCS acutely depresses
hemodynamics and grip strength in mice at doses ≥12.5 mg/kg i.p., and a concentration ≥0.52 μM in water compromises swimming
performance in larval fathead minnow. In isolated ventricular cardiomyocytes, skeletal myotubes, and adult flexor digitorum
brevis fibers TCS depresses electrically evoked ECC within ∼10–20 min. In myotubes, nanomolar to low micromolar TCS initially
potentiates electrically evoked Ca2+ transients followed by complete failure of ECC, independent of Ca2+ store depletion or block of RyR1 channels. TCS also completely blocks excitation-coupled Ca2+ entry. Voltage clamp experiments showed that TCS partially inhibits L-type Ca2+ currents of cardiac and skeletal muscle, and [3H]PN200 binding to skeletal membranes is noncompetitively inhibited by TCS in the same concentration range that enhances [3H]ryanodine binding. TCS potently impairs orthograde and retrograde signaling between L-type Ca2+ and RyR channels in skeletal muscle, and L-type Ca2+ entry in cardiac muscle, revealing a mechanism by which TCS weakens cardiac and skeletal muscle contractility in a manner
that may negatively impact muscle health, especially in susceptible populations.
Co-reporter:Bora Inceoglu;Karen M. Wagner;Jun Yang;Ahmed Bettaieb;Nils H. Schebb;Sung Hee Hwang;Christophe Morisseau;Fawaz G. Haj
PNAS 2012 109 (28 ) pp:11390-11395
Publication Date(Web):2012-07-10
DOI:10.1073/pnas.1208708109
The nerve damage occurring as a consequence of glucose toxicity in diabetes leads to neuropathic pain, among other problems.
This pain dramatically reduces the quality of life in afflicted patients. The progressive damage to the peripheral nervous
system is irreversible although strict control of hyperglycemia may prevent further damage. Current treatments include tricyclic
antidepressants, anticonvulsants, and opioids, depending on the severity of the pain state. However, available therapeutics
have drawbacks, arguing for the need to better understand the pathophysiology of neuropathic pain and develop novel treatments.
Here we demonstrate that stabilization of a class of bioactive lipids, epoxygenated fatty acids (EpFAs), greatly reduces allodynia
in rats caused by streptozocin-induced type I diabetes. Inhibitors of the soluble epoxide hydrolase (sEHI) elevated and stabilized
the levels of plasma and spinal EpFAs, respectively, and generated dose-dependent antiallodynic effects more potently and
efficaciously than gabapentin. In acute experiments, positive modulation of EpFAs did not display differences in insulin sensitivity,
glucose tolerance, or insulin secretion, indicating the efficacy of sEHIs are not related to the glycemic status. Quantitative
metabolomic analysis of a panel of 26 bioactive lipids demonstrated that sEHI-mediated antiallodynic effects coincided with
a selective elevation of the levels of EpFAs in the plasma, and a decrease in degradation products coincided with the dihydroxy
fatty acids in the spinal cord. Overall, these results argue that further efforts in understanding the spectrum of effects
of EpFAs will yield novel opportunities in treating neuropathic pain.
Co-reporter:Timo Frömel;Benno Jungblut;Caroline Trouvain;Stefanie Dimmeler;Eduardo Barbosa-Sicard;Jiong Hu;Stefan Liebner;Rüdiger Popp;Ingrid Fleming
PNAS 2012 Volume 109 (Issue 25 ) pp:9995-10000
Publication Date(Web):2012-06-19
DOI:10.1073/pnas.1206493109
Fatty acid epoxides are important lipid signaling molecules involved in the regulation of vascular tone and homeostasis. Tissue
and plasma levels of these mediators are determined by the activity of cytochrome P450 epoxygenases and the soluble epoxide
hydrolase (sEH), and targeting the latter is an effective way of manipulating epoxide levels in vivo. We investigated the
role of the sEH in regulating the mobilization and proliferation of progenitor cells with vasculogenic/reparative potential.
Our studies revealed that sEH down-regulation/inhibition impaired the development of the caudal vein plexus in zebrafish,
and decreased the numbers of lmo2/cmyb-positive progenitor cells therein. In mice sEH inactivation attenuated progenitor cell
proliferation (spleen colony formation), but the sEH products 12,13-dihydroxyoctadecenoic acid (12,13-DiHOME) and 11,12- dihydroxyeicosatrienoic
acid stimulated canonical Wnt signaling and rescued the effects of sEH inhibition. In murine bone marrow, the epoxide/diol
content increased during G-CSF–induced progenitor cell expansion and mobilization, and both mobilization and spleen colony
formation were reduced in sEH−/− mice. Similarly, sEH−/− mice showed impaired functional recovery following hindlimb ischemia, which was rescued following either the restoration
of bone marrow sEH activity or treatment with 12,13-DiHOME. Thus, sEH activity is required for optimal progenitor cell proliferation,
whereas long-term sEH inhibition is detrimental to progenitor cell proliferation, mobilization, and vascular repair.
Co-reporter:Hee-Joo Kim, Mark McCoy, Shirley J. Gee, Gualberto G. González-Sapienza, and Bruce D. Hammock
Analytical Chemistry 2011 Volume 83(Issue 1) pp:246
Publication Date(Web):December 9, 2010
DOI:10.1021/ac102353z
Immuno polymerase chain reaction (IPCR) is an analytical technology based on the excellent affinity and specificity of antibodies combined with the powerful signal amplification of polymerase chain reaction (PCR), providing superior sensitivity to classical immunoassays. Here we present a novel type of IPCR termed phage anti-immunocomplex assay real-time PCR (PHAIA-PCR) for the detection of small molecules. Our method utilizes a phage anti-immunocomplex assay (PHAIA) technology in which a short peptide loop displayed on the surface of the M13 bacteriophage binds specifically to the antibody−analyte complex, allowing the noncompetitive detection of small analytes. The phagemid DNA encoding this peptide can be amplified by PCR, and thus, this method eliminates hapten functionalization or bioconjugation of a DNA template while providing improved sensitivity. As a proof of concept, two PHAIA-PCRs were developed for the detection of 3-phenoxybenzoic acid, a major urinary metabolite of some pyrethroid insecticides, and molinate, a herbicide implicated in fish kills. Our results demonstrate that phage DNA can be a versatile material for IPCR development, enabling universal amplification when the common element of the phagemid is targeted or specific amplification when the real time PCR probe is designed to anneal the DNA encoding the peptide. The PHAIA-PCRs proved to be 10-fold more sensitive than conventional PHAIA and significantly faster using magnetic beads for rapid separation of reactants. The assay was validated with both agricultural drain water and human urine samples, showing its robustness for rapid monitoring of human exposure or environmental contamination.
Co-reporter:Sung Hee Hwang ; Karen M. Wagner ; Christophe Morisseau ; Jun-Yan Liu ; Hua Dong ; Aaron T. Wecksler
Journal of Medicinal Chemistry 2011 Volume 54(Issue 8) pp:3037-3050
Publication Date(Web):March 24, 2011
DOI:10.1021/jm2001376
A series of dual inhibitors containing a 1,5-diarylpyrazole and a urea were designed, synthesized, and evaluated as novel COX-2/sEH dual inhibitors in vitro using recombinant enzyme assays and in vivo using a lipopolysaccharide (LPS) induced model of pain in rats. The best inhibition potencies and selectivity for sEH and COX-2 over COX-1 were obtained with compounds (21b, 21i, and 21j) in which both the 1,5-diaryl-pyrazole group and the urea group are linked with a three-methylene group. Compound 21i showed the best pharmacokinetic profiles in both mice and rats (higher AUC and longer half-life). Following subcutaneous administration at 10 mg/kg, compound 21i exhibited antiallodynic activity that is more effective than the same dose of either a COX-2 inhibitor (celecoxib) or a sEH inhibitor (t-AUCB) alone, as well as coadministration of both inhibitors. Thus, these novel dual inhibitors exhibited enhanced in vivo antiallodynic activity in a nociceptive behavioral assay.
Co-reporter:Nils Helge Schebb, Bora Inceoglu, Ki Chang Ahn, Christophe Morisseau, Shirley J. Gee, and Bruce D. Hammock
Environmental Science & Technology 2011 Volume 45(Issue 7) pp:3109-3115
Publication Date(Web):March 7, 2011
DOI:10.1021/es103650m
The antibacterial soap additive triclocarban (TCC) is widely used in personal care products. TCC has a high environmental persistence. We developed and validated a sensitive online solid-phase extraction−LC−MS/MS method to rapidly analyze TCC and its major metabolites in urine and other biological samples to assess human exposure. We measured human urine concentrations 0−72 h after showering with a commercial bar soap containing 0.6% TCC. The major route of renal elimination was excretion as N-glucuronides. The absorption was estimated at 0.6% of the 70 ± 15 mg of TCC in the soap used. The TCC-N-glucuronide urine concentration varied widely among the subjects, and continuous daily use of the soap led to steady state levels of excretion. In order to assess potential biological effects arising from this exposure, we screened TCC for the inhibition of human enzymes in vitro. We demonstrate that TCC is a potent inhibitor of the enzyme soluble epoxide hydrolase (sEH), whereas TCC’s major metabolites lack strong inhibitory activity. Topical administration of TCC at similar levels to rats in a preliminary in vivo study, however, failed to alter plasma biomarkers of sEH activity. Overall the analytical strategy described here revealed that use of TCC soap causes exposure levels that warrant further evaluation.
Co-reporter:Ki Chang Ahn, Hee-Joo Kim, Mark R. McCoy, Shirley J. Gee, and Bruce D. Hammock
Journal of Agricultural and Food Chemistry 2011 Volume 59(Issue 7) pp:2792-2802
Publication Date(Web):November 24, 2010
DOI:10.1021/jf1033569
This paper describes some of the early work on pyrethroid insecticides in the Casida laboratory and briefly reviews the development and application of immunochemical approaches for the detection of pyrethroid insecticides and their metabolites for monitoring environmental and human exposure. Multiple technologies can be combined to enhance the sensitivity and speed of immunochemical analysis. The pyrethroid assays are used to illustrate the use of some of these immunoreagents such as antibodies, competitive mimics, and novel binding agents such as phage-displayed peptides. The paper also illustrates reporters such as fluorescent dyes, chemiluminescent compounds, and luminescent lanthanide nanoparticles, as well as the application of magnetic separation, and automatic instrumental systems, biosensors, and novel immunological technologies. These new technologies alone and in combination result in an improved ability to both determine if effective levels of pyrethroids are being used in the field and evaluate possible contamination.
Co-reporter:Karen Wagner, Bora Inceoglu, Sarjeet S. Gill, and Bruce D. Hammock
Journal of Agricultural and Food Chemistry 2011 Volume 59(Issue 7) pp:2816-2824
Publication Date(Web):October 19, 2010
DOI:10.1021/jf102559q
The soluble epoxide hydrolase (sEH) enzyme was discovered while investigating the metabolism of xenobiotic compounds in the Casida laboratory. However, an endogenous role of sEH is to regulate the levels of a group of potent bioactive lipids, epoxygenated fatty acids (EFAs), that have pleiotropic biological activities. The EFAs, in particular the arachidonic acid derived epoxy eicosatrienoic acids (EETs), are established autocrine and paracrine messengers. The most recently discovered outcome of inhibition of sEH and increased EFAs is their effects on the sensory system and in particular their ability to reduce pain. The inhibitors of sEH block both inflammatory and neuropathic pain. Elevation of EFAs, in both the central and peripheral nervous systems, blocks pain. Several laboratories have now published a number of potential mechanisms of action for the pain-reducing effects of EFAs. This paper provides a brief history of the discovery of the sEH enzyme and argues that inhibitors of sEH through several independent mechanisms display pain-reducing effects.
Co-reporter:Nils Helge Schebb, Bora Inceoglu, Tristan Rose, Karen Wagner and Bruce D. Hammock
Analytical Methods 2011 vol. 3(Issue 2) pp:420-428
Publication Date(Web):22 Dec 2010
DOI:10.1039/C0AY00714E
High-throughput analyses of a large number of samples for pharmacokinetic (PK) studies are essential in drug development. Analysis of drug candidates from blood using LC-ESI-MS generally requires separation of the plasma fraction followed by various offline sample preparation procedures. This step is a bottleneck that impedes throughput. In order to overcome this difficulty and accelerate analysis in PK and other studies, we developed an approach allowing the direct analysis of low volumes of whole blood (10 µL) after dilution and centrifugation. Samples were injected in an online-SPE-LC-ESI-MS/MS setup allowing a total run time of only 126 s for a full gradient separation. Analytes were extracted from the matrix within 30 s by turbulent flow chromatography. Subsequently, a full gradient separation was carried out within 1.5 minutes on a 50 × 2.1 mm (1.7 µm) RP-18 column and the analytes were sensitively detected by ESI-MS/MS in SRM mode. The performance of this new ultra fast online SPE-LC-ESI-MS/MS approach was demonstrated by the analysis of diclofenac (DCF), a widely used anti-inflammatory drug. DCF eluted at stable retention times (±0.33%) with narrow peak width (FWHM 3.3 ± 0.15 s). The method displays excellent analytical performance, with a limit of detection of 6 fmol on column, a linear range of over four orders of magnitude and a negligible carry over of 0.12 ± 0.03% for DCF. The PK profile of DCF administered by topical and intraperitoneal routes in rats and by oral route in one human volunteer is investigated using this method. Finally, general applicability of the approach for drugs is demonstrated by analysis of rofecoxib and several inhibitors of the soluble epoxide hydrolase. This new method requires only readily available, off the shelf standard LC instrumentation, and is compliant with the requirements of green analytical chemistry.
Co-reporter:Guang-Jong Shieh;Hsin-Chen Liu;Ayala Luria;Ahmed Bettaieb;Yannan Xi;John D. Imig;Hiromi Inoue;Hsing-Ju Tsai;Fawaz G. Haj
PNAS 2011 Volume 108 (Issue 22 ) pp:9038-9043
Publication Date(Web):2011-05-31
DOI:10.1073/pnas.1103482108
Visceral obesity has been defined as an important element of the metabolic syndrome and contributes to the development of
insulin resistance and cardiovascular disease. Increasing endogenous levels of epoxyeicosatrienoic acids (EETs) are known
for their analgesic, antihypertensive, and antiinflammatory effects. The availability of EETs is limited primarily by the
soluble epoxide hydrolase (sEH, EPHX2), which metabolizes EETs to their less active diols. In this study, we tested the hypothesis that EETs are involved in glucose
regulation and in retarding the development of insulin resistance. To address the role of EETs in regulating glucose homeostasis
and insulin signaling, we used mice with targeted gene deletion of sEH (Ephx2-null mice) and a subsequent study with a selective sEH inhibitor. When wild-type mice are fed a high fat diet, insulin resistance
develops. However, knockout or inhibition of sEH activity resulted in a significant decrease in plasma glucose. These findings
are characterized by enhancement of tyrosyl phosphorylation of the insulin receptor, insulin receptor substrate 1, and their
downstream cascade. In addition, pancreatic islets were larger when sEH was disrupted. This effect was associated with an
increase in vasculature. These observations were supported by pharmacological inhibition of sEH. These data suggest that an
increase in EETs due to sEH-gene knockout leads to an increase in the size of islets and improved insulin signaling and sensitivity.
Co-reporter:Bora Inceoglu;Karen Wagner;Tristan Rose;Steven L. Jinks;Nils H. Schebb;Christophe Morisseau;Christine Hegedus;Robert Brosnan;Arzu Ulu
PNAS 2011 Volume 108 (Issue 12 ) pp:5093-5097
Publication Date(Web):2011-03-22
DOI:10.1073/pnas.1101073108
Pain is a major health concern even though numerous analgesic agents are available. Side effects and lack of wide-spectrum
efficacy of current drugs justify efforts to better understand pain mechanisms. Stabilization of natural epoxy-fatty acids
(EFAs) through inhibition of the soluble epoxide hydrolase (sEH) reduces pain. However, in the absence of an underlying painful
state, inhibition of sEH is ineffective. Surprisingly, a pain-mediating second messenger, cAMP, interacts with natural EFAs
and regulates the analgesic activity of sEH inhibitors. Concurrent inhibition of sEH and phosphodiesterase (PDE) dramatically
reduced acute pain in rodents. Our findings demonstrate a mechanism of action of cAMP and EFAs in the pathophysiology of pain.
Furthermore, we demonstrate that inhibition of various PDE isozymes, including PDE4, lead to significant increases in EFA
levels through a mechanism independent of sEH, suggesting that the efficacy of commercial PDE inhibitors could result in part
from increasing EFAs. The cross-talk between the two major pathways—one mediated by cAMP and the other by EFAs—paves the way
to new approaches to understand and control pain.
Co-reporter:Nils Helge Schebb;Marion Huby
Analytical and Bioanalytical Chemistry 2011 Volume 400( Issue 5) pp:
Publication Date(Web):2011 May
DOI:10.1007/s00216-011-4861-2
Soluble epoxide hydrolase (sEH) is a promising therapeutic target for the treatment of hypertension, pain, and inflammation-related diseases. In order to enable the development of sEH inhibitors (sEHIs), assays are needed for determination of their potency. Therefore, we developed a new method utilizing an epoxide of arachidonic acid (14(15)-EpETrE) as substrate. Incubation samples were directly injected without purification into an online solid phase extraction (SPE) liquid chromatography electrospray ionization tandem mass spectrometry (LC–ESI–MS–MS) setup allowing a total run time of only 108 s for a full gradient separation. Analytes were extracted from the matrix within 30 s by turbulent flow chromatography. Subsequently, a full gradient separation was carried out on a 50X2.1 mm RP-18 column filled with 1.7 μm core–shell particles. The analytes were detected with high sensitivity by ESI–MS–MS in SRM mode. The substrate 14(15)-EpETrE eluted at a stable retention time of 96 ± 1 s and its sEH hydrolysis product 14,15-DiHETrE at 63 ± 1 s with narrow peak width (full width at half maximum height: 1.5 ± 0.1 s). The analytical performance of the method was excellent, with a limit of detection of 2 fmol on column, a linear range of over three orders of magnitude, and a negligible carry-over of 0.1% for 14,15-DiHETrE. The enzyme assay was carried out in a 96-well plate format, and near perfect sigmoidal dose–response curves were obtained for 12 concentrations of each inhibitor in only 22 min, enabling precise determination of IC50 values. In contrast with other approaches, this method enables quantitative evaluation of potent sEHIs with picomolar potencies because only 33 pmol L−1 sEH were used in the reaction vessel. This was demonstrated by ranking ten compounds by their activity; in the fluorescence method all yielded IC50 ≤ 1 nmol L−1. Comparison of 13 inhibitors with IC50 values >1 nmol L−1 showed a good correlation with the fluorescence method (linear correlation coefficient 0.9, slope 0.95, Spearman’s rho 0.9). For individual compounds, however, up to eightfold differences in potencies between this and the fluorescence method were obtained. Therefore, enzyme assays using natural substrate, as described here, are indispensable for reliable determination of structure–activity relationships for sEH inhibition.
Co-reporter:Tristan E. Rose ; Christophe Morisseau ; Jun-Yan Liu ; Bora Inceoglu ; Paul D. Jones ; James R. Sanborn
Journal of Medicinal Chemistry 2010 Volume 53(Issue 19) pp:7067-7075
Publication Date(Web):September 2, 2010
DOI:10.1021/jm100691c
1,3-Disubstituted ureas possessing a piperidyl moiety have been synthesized to investigate their structure−activity relationships as inhibitors of the human and murine soluble epoxide hydrolase (sEH). Oral administration of 13 1-aryl-3-(1-acylpiperidin-4-yl)urea inhibitors in mice revealed substantial improvements in pharmacokinetic parameters over previously reported 1-adamantylurea based inhibitors. For example, 1-(1-(cyclopropanecarbonyl)piperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea (52) showed a 7-fold increase in potency, a 65-fold increase in Cmax, and a 3300-fold increase in AUC over its adamantane analogue 1-(1-adamantyl)-3-(1-propionylpiperidin-4-yl)urea (2). This novel sEH inhibitor showed a 1000-fold increase in potency when compared to morphine by reducing hyperalgesia as measured by mechanical withdrawal threshold using the in vivo carrageenan induced inflammatory pain model.
Co-reporter:Shizuo G. Kamita, Mark D. Wogulis, Christopher S. Law, Christophe Morisseau, Hiromasa Tanaka, Huazhang Huang, David K. Wilson and Bruce D. Hammock
Biochemistry 2010 Volume 49(Issue 17) pp:
Publication Date(Web):March 22, 2010
DOI:10.1021/bi901641x
Juvenile hormone (JH) is a key insect developmental hormone that is found at low nanomolar levels in larval insects. The methyl ester of JH is hydrolyzed in many insects by an esterase that shows high specificity for JH. We have previously determined a crystal structure of the JH esterase (JHE) of the tobacco hornworm Manduca sexta (MsJHE) [Wogulis, M., Wheelock, C. E., Kamita, S. G., Hinton, A. C., Whetstone, P. A., Hammock, B. D., and Wilson, D. K. (2006) Biochemistry 45, 4045−4057]. Our molecular modeling indicates that JH fits very tightly within the substrate binding pocket of MsJHE. This tight fit places two noncatalytic amino acid residues, Phe-259 and Thr-314, within the appropriate distance and geometry to potentially interact with the α,β-unsaturated ester and epoxide, respectively, of JH. These residues are highly conserved in numerous biologically active JHEs. Kinetic analyses of mutants of Phe-259 or Thr-314 indicate that these residues contribute to the low KM that MsJHE shows for JH. This low KM, however, comes at the cost of reduced substrate turnover. Neither nucleophilic attack of the resonance-stabilized ester by the catalytic serine nor the availability of a water molecule for attack of the acyl-enzyme intermediate appears to be a rate-determining step in the hydrolysis of JH by MsJHE. We hypothesize that the release of the JH acid metabolite from the substrate binding pocket limits the catalytic cycle. Our findings also demonstrate that chemical bond strength does not necessarily correlate with how reactive the bond will be to metabolism.
Co-reporter:Hee-Joo Kim, Martin A. Rossotti, Ki Chang Ahn, Gualberto G. González-Sapienza, Shirley J. Gee, Ruthie Musker, Bruce D. Hammock
Analytical Biochemistry 2010 Volume 401(Issue 1) pp:38-46
Publication Date(Web):1 June 2010
DOI:10.1016/j.ab.2010.01.040
We present a new application of the noncompetitive phage anti-immunocomplex assay (PHAIA) by converting an existing competitive assay to a versatile noncompetitive sandwich-type format using immunocomplex binding phage-borne peptides to detect the brominated flame retardant, brominated diphenyl ether 47 (BDE 47). Three phage-displayed 9-mer disulfide-constrained peptides that recognize the BDE 47–polyclonal antibody immunocomplex were isolated. The resulting PHAIAs showed variable sensitivities, and the most sensitive peptide had a dose–response curve with an SC50 (concentration of analyte producing 50% saturation of the signal) of 0.7 ng/ml BDE 47 and a linear range of 0.3–2 ng/ml, which was nearly identical to the best heterologous competitive format (IC50 of 1.8 ng/ml, linear range of 0.4–8.5/ml). However, the PHAIA was 1400-fold better than homologous competitive assay. The validation of the PHAIA with extracts of house furniture foam as well as human and calf sera spiked with BDE 47 showed overall recovery of 80–113%. The PHAIA was adapted to a dipstick format (limit of detection of 3.0 ng/ml), and a blind test with six random extracts of local house furniture foams showed that the results of the PHAIA and dipstick assay were consistent, giving the same positive and negative detection.
Co-reporter:Yi-Xin Jim Wang;Arzu Ulu;Le-Ning Zhang;Bruce Hammock
Current Atherosclerosis Reports 2010 Volume 12( Issue 3) pp:174-183
Publication Date(Web):2010 May
DOI:10.1007/s11883-010-0108-5
Like many eicosanoids, epoxyeicosatrienoic acids (EETs) have multiple biological functions, including reduction of blood pressure, inflammation, and atherosclerosis in multiple species. Hydration of EETs by the soluble epoxide hydrolase (sEH) is the major route of their degradation to the less bioactive diols. Inhibition of the sEH stabilizes EETs, thus, enhancing the beneficial effects of EETs. Human data show an association of sEH (Ephx2) gene polymorphisms with increased risk of atherosclerosis and cardiovascular diseases. These data suggest a potential therapeutic effect of sEH inhibitors (sEHI) in the treatment of atherosclerosis. Indeed, two laboratories reported independently that using different sEHIs in apolipoprotein E–deficient mice significantly attenuated atherosclerosis development and aneurysm formation. The antiatherosclerotic effects of sEHI are correlated with elevation in EET levels and associated with reduction of low-density lipoprotein and elevation of high-density lipoprotein cholesterols, as well as attenuation of expression of proinflammatory genes and proteins. In addition, the antihypertensive effects and improvement of endothelial function also contribute to the mechanism of the antiatherosclerotic effects of sEHI. The broad spectrum of biological action of EETs and sEHIs with multiple biological beneficial actions provides a promising new class of therapeutics for atherosclerosis and other cardiovascular diseases.
Co-reporter:Jun-Yan Liu;Jun Yang;Nan Li;Hong Qiu;Nipavan Chiamvimonvat;Yi Zhu;Ning Li;Ding Ai
PNAS 2010 Volume 107 (Issue 39 ) pp:17017-17022
Publication Date(Web):2010-09-28
DOI:10.1073/pnas.1011278107
Chronic administration of high levels of selective COX-2 inhibitors (coxibs), particularly rofecoxib, valdecoxib, and parecoxib,
increases risk for cardiovascular disease. Understanding the possibly multiple mechanisms underlying these adverse cardiovascular
events is critical for evaluating the risks and benefits of coxibs and for development of safer coxibs. The current understanding
of these mechanisms is likely incomplete. Using a metabolomics approach, we demonstrate that oral administration of rofecoxib
for 3 mo results in a greater than 120-fold higher blood level of 20-hydroxyeicosatetraenoic acid (20-HETE), which correlates
with a significantly shorter tail bleeding time in a murine model. We tested the hypothesis that this dramatic increase in
20-HETE is attributable to inhibition of its metabolism and that the shortened bleeding time following rofecoxib administration
is attributable, in part, to this increase. The s.c. infusion of 20-HETE shortened the tail bleeding time dramatically. Neither
20-HETE biosynthesis nor cytochrome P4A-like immune reactivity was increased by rofecoxib administration, but 20-HETE production
increased in vitro with the addition of coxib. 20-HETE is significantly more potent than its COX-mediated metabolites in shortening
clotting time in vitro. Furthermore, 20-HETE but not rofecoxib significantly increases rat platelet aggregation in vitro in
a dose-dependent manner. These data suggest 20-HETE as a marker of rofecoxib exposure and that inhibition of 20-HETE's degradation
by rofecoxib is a partial explanation for its dramatic increase, the shortened bleeding time, and, possibly, the adverse cardiovascular
events associated with rofecoxib.
Co-reporter:Jun Yang, Kara Schmelzer, Katrin Georgi and Bruce D. Hammock
Analytical Chemistry 2009 Volume 81(Issue 19) pp:8085
Publication Date(Web):August 28, 2009
DOI:10.1021/ac901282n
Cyclooxygenase, lipoxygenase, and epoxygenase derived oxylipins, especially eicosanoids, play important roles in many physiological processes. Assessment of oxidized fatty acid levels is important for understanding their homeostatic and pathophysiological roles. Most reported methods examine these pathways in isolation. The work described here employed a solid phase extraction-liquid chromatography-electrospray ionization MS/MS (SPE-LC-ESI MS/MS) method to monitor these metabolites. In 21 min, 39 oxylipins were quantified along with eight corresponding internal standards. The limits of quantification were between 0.07 and 32 pg (20 pM−10 nM). Finally, the validated method was used to evaluate oxylipin profiles in lipopolysaccharide-exposed mice, an established septic inflammatory model. The method described here offers a useful tool for the evaluation of complex regulatory oxylipin responses in in vitro or in vivo studies.
Co-reporter:Ki Chang Ahn, Shirley J. Gee, Hsing-Ju Tsai, Deborah Bennett, Marcia G. Nishioka, Arlene Blum, Elana Fishman and Bruce D. Hammock
Environmental Science & Technology 2009 Volume 43(Issue 20) pp:7784-7790
Publication Date(Web):September 15, 2009
DOI:10.1021/es9009037
We developed a selective competitive enzyme-linked immunosorbent assay (ELISA) to monitor environmental and human exposure to polybrominated diphenyl ether BDE-47 that is used as a flame retardant. 2,2′,4,4′-Tetrabromodiphenyl ether (BDE-47), a dominant PBDE congener of toxicological concern, was the target analyte. To achieve effective hapten presentation on the carrier protein for antibody production, immunizing haptens with a rigid double-bonded hydrocarbon linker introduced at different positions on the target molecule were synthesized as well as coating haptens that mimic a characteristic fragment of the molecule. Rabbit antisera produced against each immunizing antigen were screened against competitive hapten coating antigens. Under optimized competitive indirect ELISA conditions, the linear detection range in the assay buffer that includes 50% dimethyl sulfoxide was 0.35−8.50 μg/L with an IC50 value of 1.75 μg/L for BDE-47. Little or no cross-reactivity (<6%) was observed to related PBDE congeners containing the BDE-47 moiety and other halogenated compounds. Using a magnetic particle-based competitive direct ELISA increased the sensitivity by 10-fold over the indirect ELISA. The ELISA provided quantitative results when performed on small volume/weight samples such as dust, furniture foam, and blood/serum following sample preparation, suggesting a convenient screening tool.
Co-reporter:Jun-Yan Liu;Hsing-Ju Tsai;Sung Hee Hwang;Paul D Jones;Christophe Morisseau ;Bruce D Hammock
British Journal of Pharmacology 2009 Volume 156( Issue 2) pp:284-296
Publication Date(Web):
DOI:10.1111/j.1476-5381.2008.00009.x
Background and purpose: Early soluble epoxide hydrolase inhibitors (sEHIs) such as 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) are effective anti-hypertensive and anti-inflammatory agents in various animal models. However, their poor metabolic stability and limited water solubility make them difficult to use pharmacologically. Here we present the evaluation of four sEHIs for improved pharmacokinetic properties and the anti-inflammatory effects of one sEHI.
Experimental approach: The pharmacokinetic profiles of inhibitors were determined following p.o. (oral) administration and serial bleeding in mice. Subsequently the pharmacokinetics of trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB), the most promising inhibitor, was further studied following s.c. (subcutaneous), i.v. (intravenous) injections and administration in drinking water. Finally, the anti-inflammatory effect of t-AUCB was evaluated by using a lipopolysaccharide (LPS)-treated murine model.
Key results: Better pharmacokinetic parameters (higher Cmax, longer t1/2 and greater AUC) were obtained from the tested inhibitors, compared with AUDA. Oral bioavailability of t-AUCB (0.1 mg·kg−1) was 68 ± 22% (n = 4), and giving t-AUCB in drinking water is recommended as a feasible, effective and easy route of administration for chronic studies. Finally, t-AUCB (p.o.) reversed the decrease in plasma ratio of lipid epoxides to corresponding diols (a biomarker of soluble epoxide hydrolase inhibition) in lipopolysaccharide-treated mice. The in vivo potency of 1 mg·kg−1 of t-AUCB (p.o.) was better in this inflammatory model than that of 10 mg·kg−1 of AUDA-butyl ester (p.o) at 6 h after treatment.
Conclusions and implications: t-AUCB is a potent sEHI with improved pharmacokinetic properties. This compound will be a useful tool for pharmacological research and a promising starting point for drug development.
Co-reporter:Mikaela Nichkova, Xun Fu, Zheng Yang, Ping Zhong, James R. Sanborn, Dan Chang, Shirley J. Gee and Bruce D. Hammock
Journal of Agricultural and Food Chemistry 2009 Volume 57(Issue 13) pp:5673-5679
Publication Date(Web):June 15, 2009
DOI:10.1021/jf900652a
Pesticide residue analysis in citrus oils is very important for their quality and marketing. This study assessed the reliability and sensitivity of enzyme-linked immunosorbent assays (ELISA) for simazine and cypermethrin screening in orange oil. Simazine was analyzed after extraction of the oil with methanolic phosphate buffer with a limit of quantitation (LOQ) of 40 μg/L for 1-fold and ∼100 μg/L for 10-fold oils. Due to matrix effects the immunoanalysis of cypermethrin required hexane−acetonitrile partitioning followed by silica solid phase extraction. The method detected levels higher than 0.5 ppm (mg/L). This LOQ is lower than the U.S. EPA tolerance level (0.9 ppm) for cypermethrin in citrus oils. A good correlation (r2 0.99) between ELISA and LC-MS/MS was observed for the analysis of both analytes in 1-fold orange oil. Immunochemical screening can be used to reduce instrumental analysis costs by its use in preliminary orange oil screening.
Co-reporter:Hee-Joo Kim, Ki Chang Ahn, Andrés González-Techera, Gualberto G. González-Sapienza, Shirley J. Gee, Bruce D. Hammock
Analytical Biochemistry 2009 Volume 386(Issue 1) pp:45-52
Publication Date(Web):1 March 2009
DOI:10.1016/j.ab.2008.12.003
Noncompetitive immunoassays are advantageous over competitive assays for the detection of small molecular weight compounds. We recently demonstrated that phage peptide libraries can be an excellent source of immunoreagents that facilitate the development of sandwich-type noncompetitive immunoassays for the detection of small analytes, avoiding the technical challenges of producing anti-immunocomplex antibody. In this work we explore a new format that may help to optimize the performance of the phage anti-immunocomplex assay (PHAIA) technology. As a model system we used a polyclonal antibody to 3-phenoxybenzoic acid (3-PBA) and an anti-immunocomplex phage clone bearing the cyclic peptide CFNGKDWLYC. The assay setup with the biotinylated antibody immobilized onto streptavidin-coated magnetic beads significantly reduced the amount of coating antibody giving identical sensitivity (50% saturation of the signal (SC50) = 0.2–0.4 ng/ml) to the best result obtained with direct coating of the antibody on ELISA plates. The bead-based assay tolerated up to 10 and 5% of methanol and urine matrix, respectively. This assay system accurately determined the level of spiked 3-PBA in different urine samples prepared by direct dilution or clean-up with solid-phase extraction after acidic hydrolysis with overall recovery of 80–120%.
Co-reporter:Ding Ai;Wei Pang;Ming Xu;Nan Li;Paul D. Jones;Jun Yang;Youyi Zhang;John Y.-J. Shyy;Nipavan Chiamvimonvat;Yi Zhu
PNAS 2009 Volume 106 (Issue 2 ) pp:564-569
Publication Date(Web):2009-01-13
DOI:10.1073/pnas.0811022106
Pathophysiological cardiac hypertrophy is one of the most common causes of heart failure. Epoxyeicosatrienoic acids, hydrolyzed
and degraded by soluble epoxide hydrolase (sEH), can function as endothelium-derived hyperpolarizing factors to induce dilation
of coronary arteries and thus are cardioprotective. In this study, we investigated the role of sEH in two rodent models of
angiotensin II (Ang II)-induced cardiac hypertrophy. The protein level of sEH was elevated in the heart of both spontaneously
hypertensive rats and Ang II-infused Wistar rats. Blocking the Ang II type 1 receptor with losartan could abolish this induction.
Administration of a potent sEH inhibitor (sEHI) prevented the pathogenesis of the Ang II-induced hypertrophy, as demonstrated
by decreased left-ventricular hypertrophy assessed by echocardiography, reduced cardiomyocyte size, and attenuated expression
of hypertrophy markers, including atrial natriuretic factor and β-myosin heavy chain. Because sEH elevation was not observed
in exercise- or norepinephrine-induced hypertrophy, the sEH induction was closely associated with Ang II-induced hypertrophy.
In vitro, Ang II upregulated sEH and hypertrophy markers in neonatal cardiomyocytes isolated from rat and mouse. Expression of these
marker genes was elevated with adenovirus-mediated sEH overexpression but decreased with sEHI treatment. These results were
supported by studies in neonatal cardiomyocytes from sEH−/− mice. Our results suggest that sEH is specifically upregulated by Ang II, which directly mediates Ang II-induced cardiac
hypertrophy. Thus, pharmacological inhibition of sEH would be a useful approach to prevent and treat Ang II-induced cardiac
hypertrophy.
Co-reporter:Craig E. Wheelock, Kosuke Nishi, Andy Ying, Paul D. Jones, Michael E. Colvin, Marilyn M. Olmstead, Bruce D. Hammock
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 4) pp:2114-2130
Publication Date(Web):15 February 2008
DOI:10.1016/j.bmc.2007.10.081
Carboxylesterases metabolize numerous exogenous and endogenous ester-containing compounds including the chemotherapeutic agent CPT-11, anti-influenza viral agent oseltamivir, and many agrochemicals. Trifluoromethyl ketone (TFK)-containing compounds with a sulfur atom β to the ketone moiety are some of the most potent carboxylesterase and amidase inhibitors identified to date. This study examined the effects of alkyl chain length (i.e., steric effects) and sulfur oxidation state upon TFK inhibitor potency (IC50) and binding kinetics (ki). The selective carboxylesterase inhibitor benzil was used as a non-TFK containing control. These effects were examined using two commercial esterases (porcine and rabbit liver esterase) and two human recombinant esterases (hCE-1 and hCE-2) as well as human recombinant fatty acid amide hydrolase (FAAH). In addition, the inhibition mechanism was examined using a combination of 1H NMR, X-ray crystallography, and ab initio calculations. Overall, the data show that while sulfur oxidation state profoundly affects both inhibitor potency and binding kinetics, the steric effects dominate and override the contributions of sulfur oxidation. In addition, the data suggest that inclusion of a sulfur atom β to the ketone contributes an increase (∼5-fold) in inhibitor potency due to effects upon ketone hydration and/or intramolecular hydrogen bond formation. These results provide further information on the nature of the TFK binding interaction and will be useful in increasing our understanding of this basic biochemical process.
Co-reporter:Craig E. Wheelock, Jenny Forshed, Susumu Goto, Bruce D. Hammock and John W. Newman
Chemical Research in Toxicology 2008 Volume 21(Issue 3) pp:583
Publication Date(Web):February 6, 2008
DOI:10.1021/tx7002454
Pyridine is a prototypical inducer of cytochrome P450 (CYP) 2E1, an enzyme associated with cellular oxidative stress and membrane damage. To better understand the effect of this treatment on cellular lipids, the influence of pyridine exposure (100 mg/kg/day i.p. for 5 days) on fatty acids, fatty esters, and fatty alcohol ethers in brain, heart, liver, and adipose tissue from male Swiss Webster mice was investigated. Lipid levels in cholesterol esters, triglycerides, free fatty acids, cardiolipin, sphingomyelin, and glycerylphospholipids were quantified. Pyridine altered the level and composition of lipids involved in membrane structure (i.e., sphingomyelin, phosphatidylethanolamines, and plasmalogens), energy metabolism (i.e., free fatty acids), and long-chain fatty acid transport (i.e., cholesterol esters) in a tissue-specific manner. Subtle changes in cholesterol esters were observed in all tissues. Sphingomyelin in the brain and heart were depleted in monounsaturated fatty acids (1.4- and 1.5-fold, respectively), while the liver sphingomyelin concentrations increased (1.5-fold). Pyridine exposure also increased heart free fatty acids by 1.3-fold, enriched cardiac phosphatidylethanolamine in long-chain polyunsaturated fatty acids by 1.3-fold, and depleted cardiolipin-associated plasmalogens by 3.8-fold. Phosphatidylethanolamines in the brain were also enriched in both saturated fatty acids (1.2-fold) and polyunsaturated fatty acids (1.3-fold) but were depleted in plasmalogens (2.9-fold). In particular, the levels of phosphatidylethanolamine-associated arachidonic (AA) and docosahexaenoic acid (DHA) in both brain and cardiac tissues significantly decreased following pyridine exposure. Considering the hypothetical role of plasmalogens as membrane-bound reactive oxygen scavengers, the current findings suggest that the brain and heart should be the focus of future studies on the toxicity of pyridine, as well as other CYP 2E1 inducers.
Co-reporter:Christophe Morisseau, John W. Newman, Craig E. Wheelock, Thomas Hill III, Dexter Morin, Alan R. Buckpitt and Bruce D. Hammock
Chemical Research in Toxicology 2008 Volume 21(Issue 4) pp:951
Publication Date(Web):March 25, 2008
DOI:10.1021/tx700446u
The microsomal epoxide hydrolase (mEH) plays a significant role in the metabolism of xenobiotics such as polyaromatic toxicants. Additionally, polymorphism studies have underlined a potential role of this enzyme in relation to a number of diseases, such as emphysema, spontaneous abortion, eclampsia, and several forms of cancer. We recently demonstrated that fatty amides, such as elaidamide, represent a new class of potent inhibitors of mEH. While these compounds are very active on recombinant mEH in vitro, they are quickly inactivated in liver extracts reducing their value in vivo. We investigated the effect of structural changes on mEH inhibition potency and microsomal stability. Results obtained indicate that the presence of a small alkyl group α to the terminal amide function and a thio-ether β to this function increased mEH inhibition by an order of magnitude while significantly reducing microsomal inactivation. The addition of a hydroxyl group 9−10 carbons from the terminal amide function resulted in better inhibition potency without improving microsomal stability. The best compound obtained, 2-nonylsulfanyl-propionamide, is a competitive inhibitor of mEH with a KI of 72 nM. Furthermore, this new inhibitor significantly reduces mEH diol production in ex vivo lungs exposed to naphthalene, underlying the usefulness of the inhibitors described herein. These novel inhibitors could be valuable tools to investigate the physiological and biological roles of mEH.
Co-reporter:Bora Inceoglu;Steven L. Jinks;Arzu Ulu;Christine M. Hegedus;Katrin Georgi;Kara R. Schmelzer;Karen Wagner;Paul D. Jones;Christophe Morisseau;
Proceedings of the National Academy of Sciences 2008 105(48) pp:18901-18906
Publication Date(Web):November 21, 2008
DOI:10.1073/pnas.0809765105
During inflammation, a large amount of arachidonic acid (AA) is released into the cellular milieu and cyclooxygenase enzymes
convert this AA to prostaglandins that in turn sensitize pain pathways. However, AA is also converted to natural epoxyeicosatrienoic
acids (EETs) by cytochrome P450 enzymes. EET levels are typically regulated by soluble epoxide hydrolase (sEH), the major
enzyme degrading EETs. Here we demonstrate that EETs or inhibition of sEH lead to antihyperalgesia by at least 2 spinal mechanisms,
first by repressing the induction of the COX2 gene and second by rapidly up-regulating an acute neurosteroid-producing gene, StARD1, which requires the synchronized presence of elevated cAMP and EET levels. The analgesic activities of neurosteroids are
well known; however, here we describe a clear course toward augmenting the levels of these molecules. Redirecting the flow
of pronociceptive intracellular cAMP toward up-regulation of StARD1 mRNA by concomitantly elevating EETs is a novel path to accomplish pain relief in both inflammatory and neuropathic pain
states.
Co-reporter:Pavel A. Aronov;Laura M. Hall;Katja Dettmer
Analytical and Bioanalytical Chemistry 2008 Volume 391( Issue 5) pp:
Publication Date(Web):2008 July
DOI:10.1007/s00216-008-2095-8
Biologically active forms of vitamin D are important analytical targets in both research and clinical practice. The current technology is such that each of the vitamin D metabolites is usually analyzed by individual assay. However, current LC-MS technologies allow the simultaneous metabolic profiling of entire biochemical pathways. The impediment to the metabolic profiling of vitamin D metabolites is the low level of 1α,25-dihydroxyvitamin D3 in human serum (15–60 pg/mL). Here, we demonstrate that liquid–liquid or solid-phase extraction of vitamin D metabolites in combination with Diels–Alder derivatization with the commercially available reagent 4-phenyl-1,2,4-triazoline-3,5-dione (PTAD) followed by ultra-performance liquid chromatography (UPLC)–electrospray/tandem mass spectrometry analysis provides rapid and simultaneous quantification of 1α,25-dihydroxyvitamin D3, 1α,25-dihydroxyvitamin D2, 24R,25-dihydroxyvitamin D3, 25-hydroxyvitamin D3 and 25-hydroxyvitamin D2 in 0.5 mL human serum at a lower limit of quantification of 25 pg/mL. Precision ranged from 1.6–4.8 % and 5–16 % for 25-hydroxyvitamin D3 and 1α,25-dihydroxyvitamin D3, respectively, using solid-phase extraction.
Co-reporter:Cinzia Chiappe, Elsa Leandri, Bruce D. Hammock and Christophe Morisseau
Green Chemistry 2007 vol. 9(Issue 2) pp:162-168
Publication Date(Web):14 Nov 2006
DOI:10.1039/B612106C
Ionic liquids (ILs) offer new possibilities for epoxide hydrolase (EH) catalyzed resolution of epoxides and for synthesis of chiral 1,2-diols. Soluble EHs from cress and mouse (csEH and msEH) and microsomal EH from rat (rmEH) were tested in several ILs. For all the enzymes tested, higher enantioselectivities were obtained in [bmim][N(Tf)2] and [bmim][PF6]. The optimized amount of water for EH activity in these ILs was established. Classical problems arising from low solubility of epoxides in water or from the high tendency of the oxirane ring to undergo chemical hydrolysis were avoided using these new media.
Co-reporter:Craig E. Wheelock;Susumu Goto;John W. Newman
Metabolomics 2007 Volume 3( Issue 2) pp:137-145
Publication Date(Web):2007 June
DOI:10.1007/s11306-007-0052-8
Peroxisome proliferator activated receptor alpha (PPARα) agonists are anti-hyperlipidemic drugs that influence fatty acid combustion, phospholipid biosynthesis and lipoprotein metabolism. To evaluate impacts on other aspects of lipid metabolism, we applied targeted metabolomics to liver, heart, brain and white adipose tissue samples from male Swiss-Webster mice exposed to a 5 day, 500 mg/kg/day regimen of i.p. clofibrate. Tissue concentrations of free fatty acids and the fatty acid content of sphingomyelin, cardiolipin, cholesterol esters, triglycerides and phospholipids were quantified. Responses were tissue-specific, with changes observed in the liver > heart >> brain > adipose. These results indicate that liver saturated fatty acid-rich triglycerides feeds clofibrate-induced monounsaturated fatty acid (MUFA) synthesis, which were incorporated into hepatic phospholipids and sphingomyelin. In addition, selective enrichment of docosahexeneoic acid in the phosphatidylserine of liver (1.7-fold), heart (1.6-fold) and brain (1.5-fold) suggests a clofibrate-dependent systemic activation of phosphatidylserine synthetase 2. Furthermore, the observed ∼20% decline in cardiac sphingomyelin is consistent with activation of a sphingomeylinase with a substrate preference for polyunsaturate-containing sphingomyelin. Finally, perturbations in the liver, brain, and adipose cholesterol esters were observed, with clofibrate exposure elevating brain cholesterol arachidonyl-esters ∼20-fold. Thus, while supporting previous findings, this study has identified novel impacts of PPARα agonist exposure on lipid metabolism that should be further explored.
Co-reporter:Deliang Guo;Ding Ai;John Y.-J. Shyy;Nanping Wang;Yi Zhu;Yi Fu;Hiromasa Tanaka;Chaoshu Tang
PNAS 2007 Volume 104 (Issue 21 ) pp:9018-9023
Publication Date(Web):
DOI:10.1073/pnas.0703229104
Co-reporter:Marja E. Koivunen, Katja Dettmer, Roel Vermeulen, Berit Bakke, Shirley J. Gee, Bruce D. Hammock
Analytica Chimica Acta 2006 Volume 572(Issue 2) pp:180-189
Publication Date(Web):21 July 2006
DOI:10.1016/j.aca.2006.05.037
Elimination of interfering substances in urine by solid phase extraction (SPE) prior to analysis resulted in 10-fold improvement in the sensitivity of atrazine mercapturate (AM) enzyme-linked immunosorbent assay (ELISA) compared to previous reports. Of the two tested SPE systems, Oasis® HLB and MCX, the mixed-mode MCX gave good recoveries (82%) of AM in spiked samples measured by ELISA, whereas the reverse-phase HLB phase was not compatible with the immunochemical method. At relatively high concentrations of urinary AM (>20 ng mL−1), sample dilution was effective enough for the elimination of interfering substances. The new liquid chromatography–mass spectrometry (LC–MS) method developed for AM utilizes online-SPE with Oasis® HLB, column switching and a stable-isotope internal standard. The limit of quantification (0.05 ng mL−1) indicates improved sensitivity compared with most previously published LC–MS methods for AM. Validation of all three methods, LC–MS, ELISA + SPE and ELISA + dilution with spiked urine samples showed good correlation between the known and measured concentrations with R2 values of 0.996, 0.957 and 0.961, respectively. When a set (n = 70 plus 12 blind duplicates) of urine samples from farmers exposed to atrazine was analyzed, there was a good agreement (R2 = 0.917) between the log normalized data obtained by ELISA + SPE and LC–MS. High correlation among the data obtained by the two tested methods and the LC–MS method by the Center of Disease Control and Prevention (CDC), together with low variability among the blind duplicates, suggests that both methods reported here would be suitable for the analysis of urinary AM as a biomarker for human exposure of atrazine.
Co-reporter:Nicola M. Wolf, Christophe Morisseau, Paul D. Jones, Bertold Hock, Bruce D. Hammock
Analytical Biochemistry 2006 Volume 355(Issue 1) pp:71-80
Publication Date(Web):1 August 2006
DOI:10.1016/j.ab.2006.04.045
Mammalian soluble epoxide hydrolase (sEH) represents a highly promising new target for drug development. Chemical inhibition of this enzyme in animal models was shown to treat hypertension and vascular inflammation as well as related syndromes. Existing sEH inhibitors are relatively potent and specific. However, the low solubility and relatively fast metabolism of described sEH inhibitors make them less than therapeutically efficient, stating the need for novel inhibitor structures. Therefore, a series of α-cyanoester and α-cyanocarbonate epoxides were evaluated as potential human sEH (HsEH) substrates for the high-throughput screen (HTS) of compound libraries. (3-Phenyl-oxiranyl)-acetic acid cyano-(6-methoxy-naphthalen-2-yl)-methyl ester (PHOME), which displayed the highest aqueous stability and solubility, was selected for the development of an HTS assay with long incubation times at room temperature. Concentrations of HsEH and PHOME were optimized to ensure assay sensitivity, reliability, and reproducibility. Assay validation, which employed these optimized concentrations, resulted in good accuracy (60–100%) and high precision (<7% relative standard deviation). In addition, an overall Z′ value of 0.7 proved the system’s robustness and potential for HTS. The developed assay system will be a valuable tool to discover new structures for the therapeutic inhibition of sEH to treat various cardiovascular diseases.
Co-reporter:Christophe Morisseau, John W. Newman, Hsing-Ju Tsai, Preston A. Baecker, Bruce D. Hammock
Bioorganic & Medicinal Chemistry Letters 2006 Volume 16(Issue 20) pp:5439-5444
Publication Date(Web):15 October 2006
DOI:10.1016/j.bmcl.2006.07.073
We prepared a series of amino acid derived cyclohexyl and adamantyl ureas and tested them as inhibitors of the human soluble epoxide hydrolase, and obtained very potent compounds (KI = 15 nM) that are >10-fold more soluble than previously described sEH inhibitors. While our lead compound 2 showed low apparent bioavailability in dogs and rats, this series of compounds revealed that sEH inhibitor structures could accept large groups that could lead to better orally available drugs.
Co-reporter:Kara R. Schmelzer;Bora Inceoglu;Lukas Kubala;In-Hae Kim;Steven L. Jinks;Jason P. Eiserich
PNAS 2006 Volume 103 (Issue 37 ) pp:13646-13651
Publication Date(Web):2006-09-12
DOI:10.1073/pnas.0605908103
Combination therapies have long been used to treat inflammation while reducing side effects. The present study was designed
to evaluate the therapeutic potential of combination treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) and previously
undescribed soluble epoxide hydrolase inhibitors (sEHIs) in lipopolysaccharide (LPS)-challenged mice. NSAIDs inhibit cyclooxygenase
(COX) enzymes and thereby decrease production of metabolites that lead to pain and inflammation. The sEHIs, such as 12-(3-adamantan-1-yl-ureido)-dodecanoic
acid butyl ester (AUDA-BE), stabilize anti-inflammatory epoxy-eicosatrienoic acids, which indirectly reduce the expression
of COX-2 protein. Here we demonstrate that the combination therapy of NSAIDs and sEHIs produces significantly beneficial effects
that are additive for alleviating pain and enhanced effects in reducing COX-2 protein expression and shifting oxylipin metabolomic
profiles. When administered alone, AUDA-BE decreased protein expression of COX-2 to 73 ± 6% of control mice treated with LPS
only without altering COX-1 expression and decreased PGE2 levels to 52 ± 8% compared with LPS-treated mice not receiving any therapeutic intervention. When AUDA-BE was used in combination
with low doses of indomethacin, celecoxib, or rofecoxib, PGE2 concentrations dropped to 51 ± 7, 84 ± 9, and 91 ± 8%, respectively, versus LPS control, without disrupting prostacyclin
and thromboxane levels. These data suggest that these drug combinations (NSAIDs and sEHIs) produce a valuable beneficial analgesic
and anti-inflammatory effect while prospectively decreasing side effects such as cardiovascular toxicity.
Co-reporter:Danyan Xu;Ning Li;Yuxia He;Valeriy Timofeyev;Ling Lu;Hsing-Ju Tsai;In-Hae Kim;Dipika Tuteja;Robertino Karlo P. Mateo;Anil Singapuri;Benjamin B. Davis;Reginald Low;Nipavan Chiamvimonvat;
Proceedings of the National Academy of Sciences 2006 103(49) pp:18733-18738
Publication Date(Web):November 27, 2006
DOI:10.1073/pnas.0609158103
Sustained cardiac hypertrophy represents one of the most common causes leading to cardiac failure. There is emerging evidence
to implicate the involvement of NF-κB in the development of cardiac hypertrophy. However, several critical questions remain
unanswered. We tested the use of soluble epoxide hydrolase (sEH) inhibitors as a means to enhance the biological activities
of epoxyeicosatrienoic acids (EETs) to treat cardiac hypertrophy. sEH catalyzes the conversion of EETs to form the corresponding
dihydroxyeicosatrienoic acids. Previous data have suggested that EETs may inhibit the activation of NF-κB-mediated gene transcription.
We directly demonstrate the beneficial effects of several potent sEH inhibitors (sEHIs) in cardiac hypertrophy. Specifically,
we show that sEHIs can prevent the development of cardiac hypertrophy using a murine model of pressure-induced cardiac hypertrophy.
In addition, sEHIs reverse the preestablished cardiac hypertrophy caused by chronic pressure overload. We further demonstrate
that these compounds potently block the NF-κB activation in cardiac myocytes. Moreover, by using in vivo electrophysiologic recordings, our study shows a beneficial effect of the compounds in the prevention of cardiac arrhythmias
that occur in association with cardiac hypertrophy. We conclude that the use of sEHIs to increase the level of the endogenous
lipid epoxides such as EETs may represent a viable and completely unexplored avenue to reduce cardiac hypertrophy by blocking
NF-κB activation.
Co-reporter:Kyung-Don Kang, Paul D. Jones, Huazhang Huang, Rong Zhang, Lyudmila A. Mostovich, Craig E. Wheelock, Takaho Watanabe, Lyudmila F. Gulyaeva, Bruce D. Hammock
Analytical Biochemistry 2005 Volume 344(Issue 2) pp:183-192
Publication Date(Web):15 September 2005
DOI:10.1016/j.ab.2005.06.032
We have previously reported the synthesis of four α-cyano-containing ethers based on 2-naphthaldehyde (2-NA) as cytochrome P450 (P450) fluorescent substrates. Activity detection was based on the formation of fluorescent 2-NA following substrate hydrolysis. A major limitation of these substrates was the need to remove NADPH, a required cofactor for P450 oxidation, before measuring 2-NA fluorescence. In this article, we report the synthesis of a new series of novel P450 substrates using 6-dimethylamino-2-naphthaldehyde (6-DMANA), which has a green fluorescent emission that is well separated from the NADPH spectrum. A major advantage of the 6-DMANA substrates is that NADPH removal is not required before fluorescence detection. We used eight α-cyano ether-based substrates to determine the O-dealkylation activity of human, mouse, and rat liver microsomes. In addition, substrate activities were compared with the commercial substrate 7-ethoxyresorufin (7-ER). The catalytic turnover rates of both the 6-DMANA- and 2-NA-based substrates were in some cases threefold faster than the catalytic turnover rate of 7-ER. The 2-NA-based substrates had greater turnover than did the 6-DMANA-based substrates. Murine and rat liver microsomes prepared from animals that had been treated with various P450 inducers were used to examine for isozyme-selective turnover of the substrates. The vastly improved optical properties and synthetic flexibility of the α-cyano ether compounds suggest that they are possibly good general P450 substrates.
Co-reporter:Paul D. Jones, Nicola M. Wolf, Christophe Morisseau, Paul Whetstone, Bertold Hock, Bruce D. Hammock
Analytical Biochemistry 2005 Volume 343(Issue 1) pp:66-75
Publication Date(Web):1 August 2005
DOI:10.1016/j.ab.2005.03.041
Inhibition of the mammalian soluble epoxide hydrolase (sEH) is a promising new therapy in the treatment of disorders resulting from hypertension and vascular inflammation. A spectrophotometric assay (4-nitrophenyl-trans-2,3-epoxy-3-phenylpropyl carbonate, NEPC) is currently used to screen libraries of chemicals; however this assay lacks the required sensitivity to differentiate the most potent inhibitors. A series of fluorescent α-cyanoester and α-cyanocarbonate epoxides that produce a strong fluorescent signal on epoxide hydrolysis by both human and murine sEH were designed as potential substrates for an in vitro inhibition assay. The murine enzyme showed a broad range of specificities, whereas the human enzyme showed the highest specificity for cyano(6-methoxy-naphthalen-2-yl)methyl trans-[(3-phenyloxiran-2-yl)methyl] carbonate. An in vitro inhibition assay was developed using this substrate and recombinant enzyme. The utility of the fluorescent assay was confirmed by determining the IC50 values for a series of known inhibitors. The new IC50 values were compared with those determined by spectrophotometric NEPC and radioactive tDPPO assays. The fluorescent assay ranked these inhibitors on the basis of IC50 values, whereas the NEPC assay did not. The ranking of inhibitor potency generally agreed with that determined using the tDPPO assay. These results show that the fluorescence-based assay is a valuable tool in the development of sEH inhibitors by revealing structure–activity relationships that previously were seen only by using the costly and labor-intensive radioactive tDPPO assay.
Co-reporter:Kevin R. Smith;Kent E. Pinkerton;Takaho Watanabe;Seung Jin Ma;Theresa L. Pedersen
PNAS 2005 Volume 102 (Issue 6 ) pp:2186-2191
Publication Date(Web):2005-02-08
DOI:10.1073/pnas.0409591102
Changes in the lungs due to smoking include inflammation, epithelial damage, and remodeling of the airways. Airway inflammation
is likely to play a critical role in the genesis and progression of tobacco smoke-induced airway disease. Soluble epoxide
hydrolase (sEH) is involved in the metabolism of endogenous chemical mediators that play an important role in inflammation.
Epoxyeicosatrienoic acids (EETs) have demonstrated antiinflammatory properties, and hydrolysis of these epoxides by sEH is
known to diminish this activity. To examine whether acute tobacco smoke-induced inflammation could be reduced by a sEH inhibitor,
12-(3-adamantane-1-yl-ureido)-dodecanoic acid n-butyl ester was given by daily s.c. injection to spontaneously hypertensive rats exposed to filtered air or tobacco smoke
for a period of 3 days (6 h/day). Acute exposure to tobacco smoke significantly increased by 3.2-fold (P < 0.05) the number of cells recovered by bronchoalveolar lavage. The sEH inhibitor significantly decreased total bronchoalveolar
lavage cell number by 37% in tobacco smoke-exposed rats with significant reductions noted in neutrophils, alveolar macrophages,
and lymphocytes. A combination of sEH inhibitor and EETs was more significant in its ability to further reduce tobacco smoke-induced
inflammation compared with the sEH inhibitor alone. The sEH inhibitor led to a shift in some plasma epoxides and diols that
are consistent with the hypothetical action of these compounds. We conclude that an sEH inhibitor, in the presence or absence
of EETs, can attenuate, in part, inflammation associated with acute exposure to tobacco smoke.
Co-reporter:Koukichi Nagasaka;Josie W. Chua;Masahiko Kobayashi;Shizuo G. Kamita;Susumu Maeda;Kazuei Mita;Toru Shimada
PNAS 2005 Volume 102 (Issue 7 ) pp:2584-2589
Publication Date(Web):2005-02-15
DOI:10.1073/pnas.0409457102
Enhanced locomotory activity (ELA), such as wandering, is a normal behavior that occurs at the end of the larval stage in
lepidopteran (butterflies and moths) insects. Baculovirus infection can also induce ELA in lepidopteran larvae. The belief
is that the virus induces this behavior to increase its transmission [Goulson, D. (1997) Oecologia 109, 219–228]. Here we show that a baculovirus-encoded protein tyrosine phosphatase (PTP) gene (ptp) induces ELA that is activated by light. ELA was induced in silkworm Bombyx mori infected with the baculovirus B. mori nucleopolyhedrovirus (BmNPV) beginning at ≈3.75 days postinfection (p.i.) and continued until 4.75 days p.i. The intensity
of the ELA was dramatically reduced immediately before death at 5.25 days p.i. Light activated the intensity of the ELA by
≈3-fold, and larvae with ELA showed positive phototropism. ELA was not induced in larvae of B. mori infected with a BmNPV ptp knockout mutant (BmPTPD). However, when a silkworm-derived ptp gene (Bmptp-h) was inserted into BmPTPD, ELA was partially recovered. Bmptp-h was identified from silkworms at 2 days after the start of the natural wandering stage. The deduced amino acid sequence of
Bmptp-h showed 48.2% identify (80.7% similarity) to the deduced amino acid sequence of BmNPV ptp. On the basis of the high homology and larval stage at which Bmptp-h was isolated, we postulate that the modern baculovirus may have acquired its ptp gene from an ancestral host and that this gene was selectively maintained because it increases virus transmission.
Co-reporter:Kara R. Schmelzer;Lukas Kubala;John W. Newman;In-Hae Kim;Jason P. Eiserich
PNAS 2005 Volume 102 (Issue 28 ) pp:9772-9777
Publication Date(Web):2005-07-12
DOI:10.1073/pnas.0503279102
As of 2004, >73 million people were prescribed antiinflammatory medication. Despite the extensive number of current products,
many people still suffer from their diseases or the pharmacological properties (side effects) of the medications. Therefore,
developing therapeutic strategies to treat inflammation remains an important endeavor. Here, we demonstrate that the soluble
epoxide hydrolase (sEH) is a key pharmacologic target for treating acute systemic inflammation. Lipopolysaccharide-induced
mortality, systemic hypotension, and histologically evaluated tissue injury were substantially diminished by administration
of urea-based, small-molecule inhibitors of sEH to C57BL/6 mice. Moreover, sEH inhibitors decreased plasma levels of proinflammatory
cytokines and nitric oxide metabolites while promoting the formation of lipoxins, thus supporting inflammatory resolution.
These data suggest that sEH inhibitors have therapeutic efficacy in the treatment and management of acute inflammatory diseases.
Co-reporter:Yi Liu;Yingjia Zhang;Kara Schmelzer;Tzong-Shyuan Lee;Xiang Fang;Yi Zhu;Arthur A. Spector;Sarjeet Gill;Christophe Morisseau;John Y.-J. Shyy
PNAS 2005 102 (46 ) pp:16747-16752
Publication Date(Web):2005-11-15
DOI:10.1073/pnas.0508081102
We previously reported that laminar flow activates peroxisome proliferator-activated receptor γ (PPARγ) in vascular endothelial
cells in a ligand-dependent manner that involves phospholipase A2 and cytochrome P450 epoxygenases. In this study, we investigated
whether epoxyeicosatrienoic acids (EETs), the catalytic products of cytochrome P450 epoxygenases, are PPARγ ligands. Competition
and direct binding assays revealed that EETs bind to the ligand-binding domain of PPARγ with K
d in the μM range. In the presence of adamantyl-ureido-dodecanoic acid (AUDA), a soluble epoxide hydrolase (sEH)-specific inhibitor,
EETs increased PPARγ transcription activity in endothelial cells and 3T3-L1 preadipocytes. Inclusion of AUDA in the perfusing
media enhanced, but overexpression of sEH reduced, the laminar flow-induced PPARγ activity. Furthermore, laminar flow augmented
cellular levels of EETs but decreased sEH at the levels of mRNA, protein, and activity. Blocking PPARγ by GW9662 abolished
the EET/AUDA-mediated antiinflammatory effect, which indicates that PPARγ is an effector of EETs.
Co-reporter:Mikaela Nichkova, Eun-Kee Park, Marja E Koivunen, Shizuo G Kamita, Shirley J Gee, Jane Chuang, Jeanette M.Van Emon, Bruce D Hammock
Talanta 2004 Volume 63(Issue 5) pp:1213-1223
Publication Date(Web):8 August 2004
DOI:10.1016/j.talanta.2004.05.030
Polychlorinated dibenzo-p-dioxins (PCDDs) and polychlorinated dibenzofurans (PCDFs) are considered highly toxic contaminants and the environmental and biological monitoring of these compounds is of great concern. Immunoassays may be used as screening methods to satisfy the growing demand for rapid and low cost analysis. In this work, we describe the application of an immunoassay that uses 2,3,7-trichloro-8-methyldibenzo-p-dioxin (TMDD) as a surrogate standard for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) to sediment and human serum samples. Sample extraction and preparation methods were developed with the aim to establish the simplest, cost-effective and efficient removal of the matrix interferences in the enzyme-linked immunosorbent assay (ELISA). The overall method for sediments is based on a hexane extraction; clean up by a multilayered silica gel column and an activated carbon column; an organic solvent exchange with DMSO–Triton X-100 and ELISA measurement. The gas chromatography–high resolution mass spectrometry (GC–HRMS) validation studies (n = 13) revealed that the method is suitable for the toxic equivalents (TEQ) screening of dioxin in sediments with a method detection limit of about 100 pg g−1 dry sediment with a precision of 13–33% R.S.D. The analysis of a large number of samples originating from different sources would be required to establish more precisely the screening level, as well as the number of false positives and negatives of dioxin TEQ by the immunoassay for sediments. The immunoassay method for sediment analysis offers improvement in speed, sample throughput, and cost in comparison to GC–HRMS. Dioxins were determined in serum samples after a simple liquid–liquid extraction and solvent exchange into DMSO–Triton X-100 without further dilution. The current method (approximate method LOQ of 200 pg ml−1 serum) is not sufficiently sensitive for the determination of dioxins in serum to measure acceptable exposure limit.
Co-reporter:Craig E Wheelock, Åsa M Wheelock, Rong Zhang, Jeanette E Stok, Christophe Morisseau, Susanna E Le Valley, Carol E Green, Bruce D Hammock
Analytical Biochemistry 2003 Volume 315(Issue 2) pp:208-222
Publication Date(Web):15 April 2003
DOI:10.1016/S0003-2697(03)00002-2
Carboxylesterases hydrolyze many pharmaceuticals and agrochemicals and have broad substrate selectivity, requiring a suite of substrates to measure hydrolytic profiles. To develop new esterase substrates, a series of α-cyanoesters that yield fluorescent products upon hydrolysis was evaluated for use in carboxylesterase assays. The use of these substrates as surrogates for Type II pyrethroid hydrolysis was tested. The results suggest that these novel analogs are appropriate for the development of high-throughput assays for pyrethroid hydrolase activity. A set of human liver microsomes was then used to determine the ability of these substrates to report esterase activity across a small population. Results were compared against standard esterase substrates. A number of the esterase substrates showed correlations, demonstrating the broad substrate selectivity of these enzymes. However, for several of the substrates, no correlations in hydrolysis rates were observed, suggesting that multiple carboxylesterase isozymes are responsible for the array of substrate hydrolytic activity. These new substrates were then compared against α-naphthyl acetate and 4-methylumbelliferyl acetate for their ability to detect hydrolytic activity in both one- and two-dimensional native electrophoresis gels. Cyano-2-naphthylmethyl butanoate was found to visualize more activity than either commercial substrate. These applications demonstrate the utility of these new substrates as both general and pyrethroid-selective reporters of esterase activity.
Co-reporter:Jozsef Lango;Jie Jing;Lili Chen;Fuat Doymaz;Isaac N. Pessah;Bora Inceoglu
PNAS 2003 Volume 100 (Issue 3 ) pp:922-927
Publication Date(Web):2003-02-04
DOI:10.1073/pnas.242735499
Scorpion venom is a complex mixture of salts, small molecules, peptides, and proteins. Scorpions employ this valuable tool
in several sophisticated ways for subduing prey, deterring predators, and possibly during mating. Here, a subtle but clever
strategy of venom utilization by scorpions is reported. Scorpions secrete a small quantity of transparent venom when initially
stimulated that we propose to name prevenom. If secretion continues, a cloudy and dense venom that is white in color is subsequently
released. The prevenom contains a combination of high K+ salt and several peptides including some that block rectifying K+ channels and elicit significant pain and toxicity because of a massive local depolarization. The presence of high extracellular
K+ in the prevenom can depolarize cells and also decrease the local electrochemical gradient making it more difficult to reestablish
the resting potential. When this positive change to the K+ equilibrium potential is combined with the blockage of rectifying K+ channels, this further delays the recovery of the resting potential, causing a prolonged effect. We propose that the prevenom
of scorpions is used as a highly efficacious predator deterrent and for immobilizing small prey while conserving metabolically
expensive venom until a certain level of stimuli is reached, after which the venom is secreted.
Co-reporter:John W. Newman;Christophe Morisseau;Todd R. Harris
PNAS 2003 Volume 100 (Issue 4 ) pp:1558-1563
Publication Date(Web):2003-02-18
DOI:10.1073/pnas.0437724100
The gene EPXH2 encodes for the soluble epoxide hydrolase (sEH), an enzyme involved in the regulation of cardiovascular and renal physiology
containing two distinct domains connected via a proline-rich linker. The C-terminal domain containing the EH catalytic activity
has been well studied. In contrast, a function for the N-terminal domain, which has high homology to the haloacid dehalogenase
family of phosphatases, has not been definitively reported. In this study we describe the N-terminal domain as a functional
phosphatase unaffected by a number of classic phosphatase inhibitors. Assuming a functional association between these catalytic
activities, dihydroxy lipid phosphates were rationalized as potential endogenous substrates. A series of phosphorylated hydroxy
lipids were therefore synthesized and found to be excellent substrates for the human sEH. The best substrate tested was the
monophosphate of dihydroxy stearic acid (threo-9/10-phosphonoxy-hydroxy-octadecanoic acid) with Km = 21 ± 0.3 μM, VMax = 338 ± 12 nmol⋅min−1⋅mg−1, and kcat = 0.35 ± 0.01 s−1. Therefore dihydroxy lipid phosphates are possible candidates for the endogenous substrates of the sEH N-terminal domain,
which would represent a novel branch of fatty acid metabolism with potential signaling functions.
Co-reporter:Evgenia G Matveeva, Guomin Shan, Ian M Kennedy, Shirley J Gee, Donald W Stoutamire, Bruce D Hammock
Analytica Chimica Acta 2001 Volume 444(Issue 1) pp:103-117
Publication Date(Web):12 October 2001
DOI:10.1016/S0003-2670(01)01161-8
Pyrethroids are widely used in agriculture as insecticides. In this study, we describe a simple one-step homogeneous fluoroimmunoassay for the glycine conjugate of phenoxybenzoic acid (PBAG), a putative pyrethroid metabolite that may be used as a biomarker of exposure to pyrethroids. Quenching fluoroimmunoassay (QFIA) is based on the competition of labeled and non-labeled pesticide for binding with antibodies and the resulting calibration curve is based on the relationship between analyte concentration and fluorescence quenching of labeled pesticide by specific antibodies. We developed a QFIA for PBAG in aqueous solution using fluorescein-labeled PBAG and polyclonal antibodies. The estimated IC50 (analyte concentration giving 50% inhibition of quenching) for PBAG was 4.5 nM. The detection limit (DL) was 0.9 nM. The dynamic range of the calibration curve was 2–50 nM. The average analytical recovery obtained by applying the method to urine samples (400- or 1000-fold urine dilution) was 85–111%. This demonstrates the QFIA to be a very simple and rapid detection method for PBAG; no washing steps and no enzyme conjugates were required.
Co-reporter:Guomin Shan, Whitney R Leeman, Shirley J Gee, James R Sanborn, A.Daniel Jones, Daniel P.Y Chang, Bruce D Hammock
Analytica Chimica Acta 2001 Volume 444(Issue 1) pp:169-178
Publication Date(Web):12 October 2001
DOI:10.1016/S0003-2670(01)01159-X
Tetrachlorodibenzo-p-dioxin (TCDD) is a well-known highly toxic compound that is present in nearly all components of the global ecosystem, including air, soil, sediment, fish and humans. Dioxin analysis is equipment intensive and expensive requiring low ppt or even ppq levels of detection. A simple, rapid, cost-effective method of analysis is desired to enable researchers to explore issues involving dioxin more quickly and to make more rational regulatory decisions. A sensitive immunoassay for TCDD has been developed in this laboratory. In the present study, this assay is further optimized using a new coating antigen system and validated with biota samples by HRGC-HRMS. The I50 of current assay to 2,3,7-trichloro, 8-methyldibenzo-p-dioxin (TMDD), a surrogate standard for TCDD, is 36±6.0 ppt, and the lower detection limit (LDL) is 4 ppt in the buffer. A good agreement between immunoassay and GC/MS was achieved for fish and egg samples. To interface with the immunoassay, a rapid sample preparation method was developed for soil samples. Without any further cleanup, the soil extracts can be directly used in the ELISA with a detection limit at the low ppt level. It can be used as an on-site tool for environmental monitoring.
Co-reporter:Masahiro Miyashita;Jack M. Presley;Bruce A. Buchholz;Kit S. Lam;Young Moo Lee;John S. Vogel
PNAS 2001 Volume 98 (Issue 8 ) pp:4403-4408
Publication Date(Web):2001-04-10
DOI:10.1073/pnas.071047998
Edman degradation remains the primary method for determining the
sequence of proteins. In this study, accelerator mass spectrometry was
used to determine the N-terminal sequence of glutathione
S-transferase at the attomole level with zeptomole
precision using a tracer of 14C. The transgenic transferase
was labeled by growing transformed Escherichia coli on
[14C]glucose and purified by microaffinity
chromatography. An internal standard of peptides on a solid phase
synthesized to release approximately equal amounts of all known amino
acids with each cycle were found to increase yield of gas phase
sequencing reactions and subsequent semimicrobore HPLC as did a
lactoglobulin carrier. This method is applicable to the sequencing of
proteins from cell culture and illustrates a path to more general
methods for determining N-terminal sequences with high sensitivity.
Co-reporter:Guomin Shan;Wei Huang;Shirley J. Gee;Bruce A. Buchholz;John S. Vogel
PNAS 2000 Volume 97 (Issue 6 ) pp:2445-2449
Publication Date(Web):2000-03-14
DOI:10.1073/pnas.040575997
The practice of immunoassay has experienced a widespread transition
from radioisotopic labeling to nonisotopic labeling over the last two
decades. Radioisotope labels have drawbacks that hamper their
applications: (i) perceived radiation hazards of
reagents, (ii) regulatory requirements and disposal
problems of working with radioactive materials, and
(iii) short shelf-life of the labeled
reagents. The advantage of isotopic labeling is the incorporation into
analytes without altering structure or reactivity, as is often the case
with ELISA or fluorescent detection systems. We developed a format for
isotope label immunoassay with the long-life isotope 14C as
the label and accelerator mass spectrometer (AMS) as the detection
system. AMS quantifies attomole levels of several isotopes, including
14C. With this exquisite sensitivity, the sensitivity of an
immunoassay is limited by the Kd of the
antibody and not the detection system. The detection limit of the
assays for atrazine and
2,3,7,8-tetrachlorodibenzo-p-dioxin was 2.0 ×
10−10 M and 2.0 × 10−11 M,
respectively, approximately an order of magnitude below the standard
enzyme immunoassay. Notably, <1 dpm (0.45 pCi) of
14C-labeled compound was used in each assay, which is well
below the limit of disposal (50 nCi per g) as nonradioactive waste.
Thus, endogenous reporter ligands quantified by AMS provide
the advantages of an RIA without the associated problems of radioactive
waste.
Co-reporter:Yoshiaki Nakagawa;Martin Sadilek;Elisabeth Lehmberg;Rafael Herrmann;Revital Herrmann;Haim Moskowitz;Young Moo Lee;Beth Ann Thomas;Ryo Shimizu;Masataka Kuroda;A. Daniel Jones
Archives of Insect Biochemistry and Physiology 1998 Volume 38(Issue 2) pp:
Publication Date(Web):6 DEC 1998
DOI:10.1002/(SICI)1520-6327(1998)38:2<53::AID-ARCH1>3.0.CO;2-W
As recombinant viruses expressing scorpion toxins are moving closer toward the market, it is important to obtain large amounts of pure toxin for biochemical characterization and the evaluation of biological activity in nontarget organisms. In the past, we purified a large amount of Androctonus australis anti-insect toxin (AaIT) present in the venom of A. australis with an analytical reversed-phase column by repeated runs of crude sample. We now report 20 times improved efficiency and speed of the purification by employing a preparative reversed-phase column. In just two consecutive HPLC steps, almost 1 mg of AaIT was obtained from 70 mg crude venom. Furthermore, additional AaIT was obtained from side fractions in a second HPLC run. Recently discovered insect selective toxin, AaIT5, was isolated simultaneously from the same venom batch. It shows different biological toxicity symptoms than the known excitatory and depressant insect toxins. AaIT5 gave 100% mortality with a dose of less than 1.3 μg against fourth-instar tobacco budworms Heliothis virescens 24 h after injection. During the purification process, we implemented mass spectrometry in addition to bioassays to monitor the presence of AaIT and AaIT5 in the HPLC fractions. Mass spectrometric screening can unambiguously follow the purification process and can greatly facilitate and expedite the downstream purification of AaIT and AaIT5 eliminating the number of bioassays required. Further, electrospray ionization was compared with matrix-assisted desorption/ionization and evaluated as a method of choice for mass spectrometric characterization of fractions from the venom purification for it provided higher mass accuracy and relative quantitation capability. Molecular models were built for AaIT5, excitatory toxin AaIT4, and depressant toxin LqhIT2. Three-dimensional structure of AaIT5 was compared with structures of the other two toxins, suggesting that AaIT5 is similar to depressant toxins. Arch. Insect Biochem. Physiol. 38:53–65, 1998. © 1998 Wiley-Liss, Inc.
Co-reporter:Jiawen Xu, Christophe Morisseau, Jun Yang, Kin Sing Stephen Lee, Shizuo G. Kamita, Bruce D. Hammock
Insect Biochemistry and Molecular Biology (September 2016) Volume 76() pp:62-69
Publication Date(Web):1 September 2016
DOI:10.1016/j.ibmb.2016.06.011
•Ingestion of an EH inhibitor (AUDA) increased the concentration of epoxy fatty acids in the midgut of the mosquito.•The ingestion also triggered a transient expression of several immune genes.•The growth of midgut bacteria was affected by the ingestion of AUDA.Epoxide hydrolases (EHs) are enzymes that play roles in metabolizing xenobiotic epoxides from the environment, and in regulating lipid signaling molecules, such as juvenile hormones in insects and epoxy fatty acids in mammals. In this study we fed mosquitoes with an epoxide hydrolase inhibitor AUDA during artificial blood feeding, and we found the inhibitor increased the concentration of epoxy fatty acids in the midgut of female mosquitoes. We also observed ingestion of AUDA triggered early expression of defensin A, cecropin A and cecropin B2 at 6 h after blood feeding. The expression of cecropin B1 and gambicin were not changed more than two fold compared to controls. The changes in gene expression were transient possibly because more than 99% of the inhibitor was metabolized or excreted at 42 h after being ingested. The ingestion of AUDA also affected the growth of bacteria colonizing in the midgut, but did not affect mosquito longevity, fecundity and fertility in our laboratory conditions. When spiked into the blood, EpOMEs and DiHOMEs were as effective as the inhibitor AUDA in reducing the bacterial load in the midgut, while EETs rescued the effects of AUDA. Our data suggest that epoxy fatty acids from host blood are immune response regulators metabolized by epoxide hydrolases in the midgut of female mosquitoes, inhibition of which causes transient changes in immune responses, and affects growth of microbes in the midgut.Download high-res image (149KB)Download full-size image
Co-reporter:Karen Wagner, Jun Yang, Bora Inceoglu, Bruce D. Hammock
The Journal of Pain (September 2014) Volume 15(Issue 9) pp:907-914
Publication Date(Web):1 September 2014
DOI:10.1016/j.jpain.2014.05.008
Highlights•Inhibiting the soluble epoxide hydrolase (sEH) is effective against neuropathic pain.•sEH inhibitors are effective against chronic pain in an operant nociceptive assay.•The small molecule sEH inhibitors show potent analgesia without reward side effects.Neuropathic pain is currently an insufficiently treated clinical condition. There remains a critical need for efficacious therapies without severe side effects to treat the uniquely persistent and tonic pain of neuropathy. Inhibitors of the soluble epoxide hydrolase (sEH) enzyme that stabilize endogenous epoxy fatty acids have demonstrated antihyperalgesia in clinical chronic inflammatory pain and modeled neuropathic pain. Recently, the conditioned place preference assay has been used to evaluate the tonic nature of neuropathy in several animal models. The current experiments use the conditioned place preference assay alongside withdrawal thresholds to investigate the antihyperalgesic efficacy of sEH inhibitors in a murine model of diabetic neuropathy. Here, the sEH inhibitor trans-4-[4-(3-trifluoromethoxyphenyl-1-ureido)-cyclohexyloxy]-benzoic acid (t-TUCB) at 10 mg/kg induced a robust place preference in diabetic neuropathic mice representative of pain relief. Importantly, this effect was absent both in control mice and in sEH-knockout mice at the same dose, indicating that t-TUCB is not positively reinforcing or rewarding. When compared to gabapentin, t-TUCB elicited a similar degree of withdrawal threshold improvement without the same degree of spontaneous locomotion decline in neuropathic mice. Overall, these experiments show that inhibiting the sEH enzyme attenuates chronic pain and offers an alternative to current side-effect-limited therapies to meet this clinical need.PerspectiveThese experiments demonstrate antihyperalgesia in a murine chronic pain model mediated by inhibiting the sEH enzyme. The results of this study indicate that inhibiting the sEH is a promising alternative for blocking chronic pain.
Co-reporter:Jiawen Xu, Christophe Morisseau, Bruce D. Hammock
Insect Biochemistry and Molecular Biology (November 2014) Volume 54() pp:42-52
Publication Date(Web):1 November 2014
DOI:10.1016/j.ibmb.2014.08.004
•An epoxide hydrolase from Anopheles gambiae has been expressed in insect cells.•The AgEH is a mammalian sEH homolog.•The AgEH shows biochemical and immunological similarities to mammalian sEHs.•Epoxy fatty acids may be chemical mediators metabolized by the AgEH in mosquitoes.•The AgEH has overlapping and complementary substrate selectivity with JHEHs.In insects, epoxide hydrolases (EHs) play critical roles in the metabolism of xenobiotic epoxides from the food resources and in the regulation of endogenous chemical mediators, such as juvenile hormones. Using the baculovirus expression system, we expressed and characterized an epoxide hydrolase from Anopheles gambiae (AgEH) that is distinct in evolutionary history from insect juvenile hormone epoxide hydrolases (JHEHs). We partially purified the enzyme by ion exchange chromatography and isoelectric focusing. The experimentally determined molecular weight and pI were estimated to be 35 kD and 6.3 respectively, different than the theoretical ones. The AgEH had the greatest activity on long chain epoxy fatty acids such as 14,15-epoxyeicosatrienoic acids (14,15-EET) and 9,10-epoxy-12Z-octadecenoic acids (9,10-EpOME or leukotoxin) among the substrates evaluated. Juvenile hormone III, a terpenoid insect growth regulator, was the next best substrate tested. The AgEH showed kinetics comparable to the mammalian soluble epoxide hydrolases, and the activity could be inhibited by AUDA [12-(3-adamantan-1-yl-ureido) dodecanoic acid], a urea-based inhibitor designed to inhibit the mammalian soluble epoxide hydrolases. The rabbit serum generated against the soluble epoxide hydrolase of Mus musculus can both cross-react with natural and denatured forms of the AgEH, suggesting immunologically they are similar. The study suggests there are mammalian sEH homologs in insects, and epoxy fatty acids may be important chemical mediators in insects.Download high-res image (56KB)Download full-size image
Co-reporter:Kelly J. Trunnelle, Deborah H. Bennett, Daniel J. Tancredi, Shirley J. Gee, Maria T. Stoecklin-Marois, Tamara E. Hennessy-Burt, Bruce D. Hammock, Marc B. Schenker
Environment International (November 2013) Volume 61() pp:57-63
Publication Date(Web):1 November 2013
DOI:10.1016/j.envint.2013.09.007
•Farm worker family pesticide use and indoor pyrethroid levels were investigated.•Outdoor pesticide use and indoor pyrethroid levels were positively associated.•Indoor pyrethroid levels and a home pesticide inventory were positively associated.Indoor pesticide exposure is a growing concern, particularly for pyrethroids, a commonly used class of pesticides. Pyrethroid concentrations may be especially high in homes of immigrant farm worker families, who often live in close proximity to agricultural fields and are faced with poor housing conditions, potentially causing high pest infestation and pesticide use. We investigate levels of pyrethroids in the house dust of farm worker family homes in a study of mothers and children living in Mendota, CA, within the population-based Mexican Immigration to California: Agricultural Safety and Acculturation (MICASA) Study. We present pesticide use data and levels of pyrethroid pesticides in indoor dust collected in 2009 as measured by questionnaires and a GC/MS analysis of the pyrethroids cis- and trans-permethrin, cypermethrin, deltamethrin, esfenvalerate and resmethrin in single dust samples collected from 55 households. Cis- and trans-permethrin had the highest detection frequencies at 67%, with median concentrations of 244 and 172 ng/g dust, respectively. Cypermethrin was detected in 52% of the homes and had a median concentration of 186 ng/g dust. Esfenvalerate, resmethrin and deltamethrin were detected in less than half the samples. We compared the pyrethroid concentrations found in our study to other studies looking at both rural and urban homes and daycares. Lower detection frequencies and/or lower median concentrations of cis- and trans-permethrin and cypermethrin were observed in our study as compared to those studies. However, deltamethrin, esfenvalerate and resmethrin were detected more frequently in the house dust from our study than in the other studies. Because households whose children had higher urinary pyrethroid metabolite levels were more likely to be analyzed in this study, a positive bias in our estimates of household pyrethroid levels may be expected. A positive association was observed with reported outdoor pesticide use and cypermethrin levels found in the indoor dust samples (rs = 0.28, p = 0.0450). There was also a positive association seen with summed pyrethroid levels in house dust and the results of a pesticide inventory conducted by field staff (rs = 0.32, p = 0.018), a potentially useful predictor of pesticide exposure in farm worker family homes. Further research is warranted to fully investigate the utility of such a measure.
Co-reporter:Karen Wagner, Bora Inceoglu, Bruce D. Hammock
Prostaglandins & Other Lipid Mediators (November 2011) Volume 96(Issues 1–4) pp:76-83
Publication Date(Web):1 November 2011
DOI:10.1016/j.prostaglandins.2011.08.001
The soluble epoxide hydrolase (sEH) enzyme regulates the levels of endogenous epoxygenated fatty acid (EFA) lipid metabolites by rapidly degrading these molecules. The EFAs have pleiotropic biological activities including the modulation of nociceptive signaling. Recent findings indicate that the EFAs, in particular the arachidonic acid (AA) derived epoxyeicosatrienoic acids (EETs), the docosahexaenoic acid (DHA) derived epoxydocosapentaenoic acids (EpDPEs) and eicosapentaenoic acid (EPA) derived epoxyeicosatetraenoic acids (EpETEs) are natural signaling molecules. The tight regulation of these metabolites speaks to their importance in regulating biological functions. In the past several years work on EFAs in regard to their activities in the nervous system evolved to demonstrate that these molecules are anti-inflammatory and anti-nociceptive. Here we focus on the recent advances in understanding the effects of sEH inhibition and increased EFAs on the nociceptive system and their ability to reduce pain. Evidence of their role in modulating pain signaling is given by their direct application and by inhibiting their degradation in various models of pain. Moreover, there is mounting evidence of EFAs role in the crosstalk between major nociceptive and anti-nociceptive systems which is reviewed herein. Overall the fundamental knowledge generated within the past decade indicates that orally bioavailable small molecule inhibitors of sEH may find a place in the treatment of a number of diverse painful conditions including inflammatory and neuropathic pain.Highlights► Epoxygenated fatty acids (EFAs), endogenous signaling molecules, mediate nociception. ► EFAs are tightly regulated primarily by the soluble epoxide hydrolase (sEH) enzyme. ► sEH inhibitors and EFAs are anti-nociceptive in inflammatory and neuropathic pain models. ► EFAs mediate crosstalk among branches in the AA cascade and among analgesic systems.
Co-reporter:Jeanette M. Van Emon, Jane C. Chuang, Robert A. Lordo, Mary E. Schrock, Mikaela Nichkova, Shirley J. Gee, Bruce D. Hammock
Chemosphere (May 2008) Volume 72(Issue 1) pp:95-103
Publication Date(Web):1 May 2008
DOI:10.1016/j.chemosphere.2008.01.012
A 96-microwell enzyme-linked immunosorbent assay (ELISA) method was evaluated to determine PCDDs/PCDFs in sediment and soil samples from an EPA Superfund site. Samples were prepared and analyzed by both the ELISA and a gas chromatography/high resolution mass spectrometry (GC/HRMS) method. Comparable method precision, accuracy, and detection level (8 ng kg−1) were achieved by the ELISA method with respect to GC/HRMS. However, the extraction and cleanup method developed for the ELISA requires refinement for the soil type that yielded a waxy residue after sample processing. Four types of statistical analyses (Pearson correlation coefficient, paired t-test, nonparametric tests, and McNemar’s test of association) were performed to determine whether the two methods produced statistically different results. The log-transformed ELISA-derived 2,3,7,8-tetrachlorodibenzo-p-dioxin values and log-transformed GC/HRMS-derived TEQ values were significantly correlated (r = 0.79) at the 0.05 level. The median difference in values between ELISA and GC/HRMS was not significant at the 0.05 level. Low false negative and false positive rates (<10%) were observed for the ELISA when compared to the GC/HRMS at 1000 ng TEQ kg−1. The findings suggest that immunochemical technology could be a complementary monitoring tool for determining concentrations at the 1000 ng TEQ kg−1 action level for contaminated sediment and soil. The ELISA could also be used in an analytical triage approach to screen and rank samples prior to instrumental analysis.
Co-reporter:El-Sayed A. El-Sheikh, Shizuo G. Kamita, Bruce D. Hammock
Pesticide Biochemistry and Physiology (March 2016) Volume 128() pp:30-36
Publication Date(Web):1 March 2016
DOI:10.1016/j.pestbp.2015.10.008
•Topical application of methoprene, fenoxycarb or pyriproxyfen significantly extends the larval stage of S. frugiperda.•In vitro JHE activity is inhibited by fenoxycarb in competition experiments with JH III.•Hemolymph JHE activity is maintained by methoprene, fenoxycarb or pyriproxyfen treatment of 6th instar S. frugiperda.Juvenile hormone analog (JHA) insecticides are biological and structural mimics of JH, a key insect developmental hormone. Toxic and anti-developmental effects of the JHA insecticides methoprene, fenoxycarb, and pyriproxyfen were investigated on the larval and pupal stages of Spodoptera littoralis and Spodoptera frugiperda. Bioassays showed that fenoxycarb has the highest toxicity and fastest speed of kill in 2nd instar S. littoralis. All three JHAs affected the development of 6th instar (i.e., final instar) and pupal S. frugiperda. JH esterase (JHE) is a critical enzyme that helps to regulate JH levels during insect development. JHE activity in the last instar S. littoralis and S. frugiperda was 11 and 23 nmol min− 1 ml− 1 hemolymph, respectively. Methoprene and pyriproxyfen showed poor inhibition of JHE activity from these insects, whereas fenoxycarb showed stronger inhibition. The inhibitory activity of fenoxycarb, however, was more than 1000-fold lower than that of OTFP, a highly potent inhibitor of JHEs. Surprisingly, topical application of methoprene, fenoxycarb or pyriproxyfen on 6th instars of S. littoralis and S. frugiperda prevented the dramatic reduction in JHE activity that was found in control insects. Our findings suggest that JHAs may function as JH agonists that play a disruptive role or a hormonal replacement role in S. littoralis and S. frugiperda.Download full-size image
Co-reporter:Shizuo G. Kamita, Kohji Yamamoto, Mary M. Dadala, Khavong Pha, Christophe Morisseau, Aurélie Escaich, Bruce D. Hammock
Insect Biochemistry and Molecular Biology (March 2013) Volume 43(Issue 3) pp:219-228
Publication Date(Web):1 March 2013
DOI:10.1016/j.ibmb.2012.12.002
Epoxide hydrolases (EHs) are α/β-hydrolase fold superfamily enzymes that convert epoxides to 1,2-trans diols. In insects EHs play critical roles in the metabolism of toxic compounds and allelochemicals found in the diet and for the regulation of endogenous juvenile hormones (JHs). In this study we obtained a full-length cDNA, hvmeh1, from the generalist feeder Heliothis virescens that encoded a highly active EH, Hv-mEH1. Of the 10 different EH substrates that were tested, Hv-mEH1 showed the highest specific activity (1180 nmol min−1 mg−1) for a 1,2-disubstituted epoxide-containing fluorescent substrate. This specific activity was more than 25- and 3900-fold higher than that for the general EH substrates cis-stilbene oxide and trans-stilbene oxide, respectively. Although phylogenetic analysis placed Hv-mEH1 in a clade with some lepidopteran JH metabolizing EHs (JHEHs), JH III was a relatively poor substrate for Hv-mEH1. Hv-mEH1 showed a unique substrate selectivity profile for the substrates tested in comparison to those of MsJHEH, a well-characterized JHEH from Manduca sexta, and hmEH, a human microsomal EH. Hv-mEH1 also showed unique enzyme inhibition profiles to JH-like urea, JH-like secondary amide, JH-like primary amide, and non-JH-like primary amide compounds in comparison to MsJHEH and hmEH. Although Hv-mEH1 is capable of metabolizing JH III, our findings suggest that this enzymatic activity does not play a significant role in the metabolism of JH in the caterpillar. The ability of Hv-mEH1 to rapidly hydrolyze 1,2-disubstituted epoxides suggests that it may play roles in the metabolism of fatty acid epoxides such as those that are commonly found in the diet of Heliothis.Graphical abstractDownload high-res image (48KB)Download full-size imageHighlights► A microsomal epoxide hydrolase (i.e., Hv-mEH1) encoding cDNA was cloned from the tobacco budworm Heliothis virescens. ► A recombinant Hv-mEH1 was expressed and its ability to hydrolyze general and fluorescent EH substrates, and juvenile hormone III (JH III) was determined. ► The ability of urea and amide compounds, including JH-like ureas and amides, to inhibit Hv-mEH1 was tested. ► Biochemical characterization suggested that JH III is a poor substrate of Hv-mEH1.
Co-reporter:Marie Hennebelle, Yurika Otoki, Jun Yang, Bruce D. Hammock, Anthony J. Levitt, Ameer Y. Taha, Walter Swardfager
Psychiatry Research (June 2017) Volume 252() pp:94-101
Publication Date(Web):1 June 2017
DOI:10.1016/j.psychres.2017.02.056
•Oxylipins are lipids that reflect regulation of inflammatory and resolution pathways.•Soluble epoxide hydrolase (sEH) produces oxylipins that are less anti-inflammatory.•sEH derived oxylipins fluctuated between mood states in seasonal depression.•sEH activity may underlie inflammatory states in symptomatic seasonal depression.•This lipidomic assay offers new potential for standardized quantitative biomarkers.Many cytochrome p450-derived lipids promote resolution of inflammation, in contrast to their soluble epoxide hydrolase(sEH)-derived oxylipin breakdown products. Here we compare plasma oxylipins and precursor fatty acids between seasons in participants with major depressive disorder with seasonal pattern (MDD-s). Euthymic participants with a history of MDD-s recruited in summer-fall were followed-up in winter. At both visits, a structured clinical interview (DSM-5 criteria) and the Beck Depression Inventory II (BDI-II) were administered. Unesterified and total oxylipin pools were assayed by liquid chromatography tandem mass-spectrometry (LC-MS/MS). Precursor fatty acids were measured by gas chromatography. In nine unmedicated participants euthymic at baseline who met depression criteria in winter, BDI-II scores increased from 4.9±4.4 to 19.9±7.7. Four sEH-derived oxylipins increased in winter compared to summer-fall with moderate to large effect sizes. An auto-oxidation product (unesterified epoxyketooctadecadienoic acid) and lipoxygenase-derived 13-hydroxyoctadecadienoic acid also increased in winter. The cytochrome p450-derived 20-COOH-leukotriene B4 (unesterified) and total 14(15)-epoxyeicosatetraenoic acid, and the sEH-derived 14,15-dihydroxyeicostrienoic acid (unesterified), decreased in winter. We conclude that winter depression was associated with changes in cytochrome p450- and sEH-derived oxylipins, suggesting that seasonal shifts in omega-6 and omega-3 fatty acid metabolism mediated by sEH may underlie inflammatory states in symptomatic MDD-s.
Co-reporter:Natalia Vasylieva, Bogdan Barnych, Debin Wan, El-Sayed A. El-Sheikh, Hai M. Nguyen, Heike Wulff, Rebecca McMahen, Mark Strynar, Shirley J. Gee, Bruce D. Hammock
Environment International (June 2017) Volume 103() pp:91-98
Publication Date(Web):1 June 2017
DOI:10.1016/j.envint.2017.03.012
•Fast and easy approach was developed for the first time synthesis of hydroxy-fipronil, a urinary metabolite of fipronil.•Hydroxy-fipronil standard was used to develop a sensitive analytical LC-MS/MS method with a LOQ of 0.4 ng/mL.•Hydroxy-fipronil is a more sensitive dose-dependent biomarker of exposure to fipronil compared to fipronil-sulfone.•Immunoassay was used for sample screening in the evaluation of exposure levels.•Hydroxyl-fipronil was tested in the insect assay and on mammalian GABAA receptors to characterize its biological activity.Occupational medical surveillance is highly desirable in manufacturing facilities where exposure to chemicals is significant. The insecticide fipronil is generally considered safe for humans but with increasing use, exposure to fipronil is of concern. Identification of urinary metabolites of fipronil may allow development of affordable, cheap and rapid procedures for human exposure evaluation. In this study we developed a fast and easy approach for synthesis of hydroxy-fipronil, a potential urinary metabolite of fipronil. This standard was used to develop a sensitive analytical LC-MS/MS method with a limit of quantification (LOQ) of 0.4 ng/mL. Fipronil sulfone, a known metabolite, and hydroxy-fipronil were quantified in urine samples from rats treated with a fipronil containing diet. Fipronil sulfone concentration centered around 20 ng/mL, while the concentration of hydroxy-fipronil was dose-dependent ranging in 10–10,000 ng/mL and thus being a more sensitive marker of fipronil exposure. A fipronil immunoassay with cross-reactivity to hydroxy-fipronil showed a good correlation in signal intensity with LC-MS data. It was also used to demonstrate the applicability of the method for sample screening in the evaluation of exposure levels.
Co-reporter:Brenna M. Flannery, Jill L. Silverman, Donald A. Bruun, Kyle R. Puhger, Mark R. McCoy, Bruce D. Hammock, Jacqueline N. Crawley, Pamela J. Lein
Neurotoxicology and Teratology (January–February 2015) Volume 47() pp:36-45
Publication Date(Web):1 January 2015
DOI:10.1016/j.ntt.2014.10.008
•Survivors of acute TETS poisoning experience persistent psychological problems.•Diazepam stops seizures in NIH Swiss mice intoxicated with lethal dose of TETS.•TETS mice do not exhibit anxiety or depression-like behavior or cognitive deficits.•Behavioral deficits seen in human TETS survivors likely due to sustained seizures.Tetramethylenedisulfotetramine (TETS) is a potent convulsant poison that is thought to trigger seizures by inhibiting the function of the type A gamma-aminobutyric acid receptor (GABAAR). Acute intoxication with TETS can cause vomiting, convulsions, status epilepticus (SE) and even death. Clinical case reports indicate that individuals who survive poisoning may exhibit long-term neuropsychological issues and cognitive deficits. Therefore, the objective of this research was to determine whether a recently described mouse model of acute TETS intoxication exhibits persistent behavioral deficits. Young adult male NIH Swiss mice received a seizure-inducing dose of TETS (0.15 mg/kg, ip) and then were rescued from lethality by administration of diazepam (5 mg/kg, ip) approximately 20 min post-TETS-exposure. TETS-intoxicated mice typically exhibited 2 clonic seizures prior to administration of diazepam with no subsequent seizures post-diazepam injection as assessed using behavioral criteria. Seizures lasted an average of 72 s. Locomotor activity, anxiety-like and depression-relevant behaviors and cognition were assessed at 1 week, 1 month and 2 months post-TETS exposure using open field, elevated-plus maze, light ↔ dark transitions, tail suspension, forced swim and novel object recognition tasks. Interestingly, preliminary validation tests indicated that NIH Swiss mice do not respond to the shock in fear conditioning tasks. Subsequent evaluation of hot plate and tail flick nociception tasks revealed that this strain exhibits significantly decreased pain sensitivity relative to age- and sex-matched C57BL/6J mice, which displayed normal contextual fear conditioning. NIH Swiss mice acutely intoxicated with TETS exhibited no significant anxiety-related, depression-relevant, learning or memory deficits relative to vehicle controls at any of the time points assessed with the exception of significantly increased locomotor activity at 2 months post-TETS intoxication. The general absence of long-term behavioral deficits in TETS-intoxicated mice on these six assays suggests that the neurobehavioral consequences of TETS exposure described in human survivors of acute TETS intoxication are likely due to sustained seizure activity, rather than a direct effect of the chemical itself. Future research efforts are directed toward developing an animal model that better recapitulates the SE and seizure duration reported in humans acutely intoxicated with TETS.
Co-reporter:Hiromasa Tanaka, Shizuo G. Kamita, Nicola M. Wolf, Todd R. Harris, Zhaoju Wu, Christophe Morisseau, Bruce D. Hammock
Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms (January 2008) Volume 1779(Issue 1) pp:
Publication Date(Web):January 2008
DOI:10.1016/j.bbagrm.2007.11.005
Soluble epoxide hydrolase (sEH) is a multifunctional protein encoded by the EPHX2 gene. The biological functions and enzyme kinetics of sEH have been extensively investigated, however, little is known about its transcriptional regulation and mechanisms of tissue specific expression. Here, a luciferase gene based reporter assay was used to identify the minimal promoter and cis regulatory elements of EPHX2. The minimal promoter was identified as a GC-rich region between nts − 374 and + 28 with respect to the putative transcriptional start site. A reporter plasmid carrying this minimal promoter showed higher or similar activities in comparison to a reporter plasmid carrying nts − 5,974 to + 28 of EPHX2 in 9 human cell lines that were tested. Sp1 binding sites that are involved in augmenting the minimal promoter activity of EPHX2 were identified by nested deletion analysis, site-specific mutation, electrophoretic mobility shift assay, and chromatin immunoprecipitation assay.
Co-reporter:Bora Inceoglu, Kara R. Schmelzer, Christophe Morisseau, Steve L. Jinks, Bruce D. Hammock
Prostaglandins & Other Lipid Mediators (January 2007) Volume 82(Issues 1–4) pp:42-49
Publication Date(Web):1 January 2007
DOI:10.1016/j.prostaglandins.2006.05.004
Early on, intriguing biological activities were found associated with the EETs using in vitro systems. Although the EETs other than the 5,6-isomer, are quite stable chemically, they are quickly degraded enzymatically with the sEH accounting in many cases for much of the metabolism. This rapid degradation often made it difficult to associate biological effects with the administration of EETs and other lipid epoxides particularly in vivo. Thus, it is the power to inhibit the sEH that has facilitated the demonstration of many physiological processes associated with EETs and possibly other epoxy fatty acids. In the last few years it has become clear that major roles of the EETs include modulation of blood pressure and modulation of inflammatory cascades. There are a number of other physiological functions now associated with the EETs including angiogenesis, neurohormone release, cell proliferation, G protein signaling, modulation of ion channel activity, and a variety of effects associated with modulation of NFκB. More recently we observed a role of the EETs as modulated by sEHI in reducing non-neuropathic pain. The array of biological effects observed with sEHI illustrates the power of modulating the degradation of chemical mediators in addition to the modulation of their biosynthesis, receptor binding and signal transduction. Many of these biological effects can be modulated by sEHIs but also by the natural eicosanoids and their mimics all of which offer therapeutic potential.
Co-reporter:Todd R. Harris, Sean Kodani, Jun Yang, Denise M. Imai, Bruce D. Hammock
The Journal of Nutritional Biochemistry (December 2016) Volume 38() pp:93-101
Publication Date(Web):1 December 2016
DOI:10.1016/j.jnutbio.2016.08.010
Exposure to the halogenated hydrocarbon carbon tetrachloride (CCl4) leads to hepatic lipid peroxidation, inflammation and fibrosis. Dietary supplementation of ω-3 fatty acids has been increasingly advocated as being generally anti-inflammatory, though its effect in models of liver fibrosis is mixed. This raises the question of whether diets high in ω-3 fatty acids will result in a greater sensitivity or resistance to liver fibrosis as a result of environmental toxicants like CCl4. In this study, we fed CCl4-treated mice a high ω-3 diet (using a mix of docosahexaenoic acid and eicosapentaenoic acid ethyl esters). We also co-administered an inhibitor of soluble epoxide hydrolase, 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU), which has been shown to boost anti-inflammatory epoxy fatty acids that are produced from both ω-3 and ω-6 dietary lipids. We showed that soluble epoxide inhibitors reduced CCl4-induced liver fibrosis. Three major results were obtained. First, the ω-3-enriched diet did not attenuate CCl4-induced liver fibrosis as judged by collagen deposition and collagen mRNA expression. Second, the ω-3-enriched diet raised hepatic tissue levels of several inflammatory lipoxygenase metabolites and prostaglandins, including PGE2. Third, treatment with TPPU in drinking water in conjunction with the ω-3-enriched diet resulted in a reduction in liver fibrosis compared to all other groups. Taken together, these results indicate that dietary ω-3 supplementation alone did not attenuate CCl4-induced liver fibrosis. Additionally, oxylipin signaling molecules may play role in the CCl4-induced liver fibrosis in the high ω-3 diet groups.
Co-reporter:Jiawen Xu, Christophe Morisseau, Jun Yang, Dadala M. Mamatha, Bruce D. Hammock
Insect Biochemistry and Molecular Biology (April 2015) Volume 59() pp:41-49
Publication Date(Web):1 April 2015
DOI:10.1016/j.ibmb.2015.02.004
•Multiple EH activities were characterized in the mosquito Culex quinquefasciatus.•Epoxy fatty acids are endogenous and xenobiotic substrates for EHs from mosquitoes.•AUDA is a useful inhibitor to investigate the biological roles of epoxy fatty acids.Culex mosquitoes have emerged as important model organisms for mosquito biology, and are disease vectors for multiple mosquito-borne pathogens, including West Nile virus. We characterized epoxide hydrolase activities in the mosquito Culex quinquefasciatus, which suggested multiple forms of epoxide hydrolases were present. We found EH activities on epoxy eicosatrienoic acids (EETs). EETs and other eicosanoids are well-established lipid signaling molecules in vertebrates. We showed EETs can be synthesized in vitro from arachidonic acids by mosquito lysate, and EETs were also detected in vivo both in larvae and adult mosquitoes by LC-MS/MS. The EH activities on EETs can be induced by blood feeding, and the highest activity was observed in the midgut of female mosquitoes. The enzyme activities on EETs can be inhibited by urea-based inhibitors designed for mammalian soluble epoxide hydrolases (sEH). The sEH inhibitors have been shown to play diverse biological roles in mammalian systems, and they can be useful tools to study the function of EETs in mosquitoes. Besides juvenile hormone metabolism and detoxification, insect epoxide hydrolases may also play a role in regulating lipid signaling molecules, such as EETs and other epoxy fatty acids, synthesized in vivo or obtained from blood feeding by female mosquitoes.Download high-res image (76KB)Download full-size image
Co-reporter:Karen Wagner, Jennifer Gilda, Jun Yang, Debin Wan, Christophe Morisseau, Aldrin V. Gomes, Bruce D. Hammock
Behavioural Brain Research (30 May 2017) Volume 326() pp:69-76
Publication Date(Web):30 May 2017
DOI:10.1016/j.bbr.2017.02.048
Co-reporter:Sean D. Kodani, Haley B. Overby, Christophe Morisseau, Jiangang Chen, Ling Zhao, Bruce D. Hammock
Toxicology Letters (16 November 2016) Volume 262() pp:92-99
Publication Date(Web):16 November 2016
DOI:10.1016/j.toxlet.2016.09.011
•Parabens inhibit the endocannabinoid enzyme fatty acid amide hydrolase (FAAH).•Paraben inhibition has time-independent, mixed-type kinetics.•Benzylparaben, the most potent paraben, inhibits FAAH with sub-micromolar potency.•Endocannabinoids may mediate paraben-enhanced adipogenesis but not through CB1 activation.Parabens are a class of small molecules that are regularly used as preservatives in a variety of personal care products. Several parabens, including butylparaben and benzylparaben, have been found to interfere with endocrine signaling and to stimulate adipocyte differentiation. We hypothesized these biological effects could be due to interference with the endocannabinoid system and identified fatty acid amide hydrolase (FAAH) as the direct molecular target of parabens. FAAH inhibition by parabens yields mixed-type and time-independent kinetics. Additionally, structure activity relationships indicate FAAH inhibition is selective for the paraben class of compounds and the more hydrophobic parabens have higher potency. Parabens enhanced 3T3-L1 adipocyte differentiation in a dose dependent fashion, different from two other FAAH inhibitors URB597 and PF622. Moreover, parabens, URB597 and PF622 all failed to enhance AEA-induced differentiation. Furthermore, rimonabant, a cannabinoid receptor 1 (CB1)-selective antagonist, did not attenuate paraben-induced adipocyte differentiation. Thus, adipogenesis mediated by parabens likely occurs through modulation of endocannabinoids, but cell differentiation is independent of direct activation of CB1 by endocannabinoids.
Co-reporter:Paul A. Whetstone, Bruce D. Hammock
Toxicon (15 March 2007) Volume 49(Issue 4) pp:576-596
Publication Date(Web):15 March 2007
DOI:10.1016/j.toxicon.2006.11.009
Since the introduction of DDT in the 1940s, arthropod pest control has relied heavily upon chemical insecticides. However, the development of insect resistance, an increased awareness of the real and perceived environmental and health impacts of these chemicals, and the need for systems with a smaller environmental footprint has stimulated the search for new insecticidal compounds, novel molecular targets, and alternative control methods. In recent decades a variety of biocontrol methods employing peptidic or proteinaceous insect-specific toxins derived from microbes, plants and animals have been examined in the laboratory and field with varying results. Among the many interdependent factors involved with the production of a cost-effective pesticide—production expense, kill efficiency, environmental persistence, pest-specificity, pest resistance-development, public perception and ease of delivery—sprayable biopesticides have not yet found equal competitive footing with chemical counterparts. However, while protein/peptide-based biopesticides continue to have limitations, advances in the technology, particularly of genetically modified organisms as biopesticidal delivery systems, has continually progressed. This review highlights the varieties of delivery methods currently practiced, examining the strengths and weaknesses of each method.
Co-reporter:Jun-Yan Liu, Hsing-Ju Tsai, Christophe Morisseau, Jozsef Lango, Sung Hee Hwang, Takaho Watanabe, In-Hae Kim, Bruce D. Hammock
Biochemical Pharmacology (15 December 2015) Volume 98(Issue 4) pp:718-731
Publication Date(Web):15 December 2015
DOI:10.1016/j.bcp.2015.10.013
Co-reporter:Sumanta Kumar Goswami, Bora Inceoglu, Jun Yang, Debin Wan, Sean D. Kodani, Carlos Antonio Trindade da Silva, Christophe Morisseau, Bruce D. Hammock
Toxicology and Applied Pharmacology (15 December 2015) Volume 289(Issue 3) pp:419-427
Publication Date(Web):15 December 2015
DOI:10.1016/j.taap.2015.10.018
•The soluble epoxide hydrolase (sEH) inhibitor TPPU is anti-hyperalgesic.•Omeprazole potentiates the anti-hyperalgesic actions of TPPU.•This potentiation is associated with increased P450 activity.•The potentiation is associated with an increase in fatty acid epoxide/diol ratio.•Joint use of sEH inhibitors and P450 inducers could result in drug–drug interactions.Epoxyeicosatrienoic acids (EETs) are potent endogenous analgesic metabolites produced from arachidonic acid by cytochrome P450s (P450s). Metabolism of EETs by soluble epoxide hydrolase (sEH) reduces their activity, while their stabilization by sEH inhibition decreases both inflammatory and neuropathic pain. Here, we tested the complementary hypothesis that increasing the level of EETs through induction of P450s by omeprazole (OME), can influence pain related signaling by itself, and potentiate the anti-hyperalgesic effect of sEH inhibitor. Rats were treated with OME (100 mg/kg/day, p.o., 7 days), sEH inhibitor TPPU (3 mg/kg/day, p.o.) and OME (100 mg/kg/day, p.o., 7 days) + TPPU (3 mg/kg/day, p.o., last 3 days of OME dose) dissolved in vehicle PEG400, and their effect on hyperalgesia (increased sensitivity to pain) induced by PGE2 was monitored. While OME treatment by itself exhibited variable effects on PGE2 induced hyperalgesia, it strongly potentiated the effect of TPPU in the same assay. The significant decrease in pain with OME + TPPU treatment correlated with the increased levels of EETs in plasma and increased activities of P450 1A1 and P450 1A2 in liver microsomes. The results show that reducing catabolism of EETs with a sEH inhibitor yielded a stronger analgesic effect than increasing generation of EETs by OME, and combination of both yielded the strongest pain reducing effect under the condition of this study.
Co-reporter:Todd R. Harris, Pavel A. Aronov, Paul D. Jones, Hiromasa Tanaka, Michael Arand, Bruce D. Hammock
Archives of Biochemistry and Biophysics (15 April 2008) Volume 472(Issue 2) pp:
Publication Date(Web):15 April 2008
DOI:10.1016/j.abb.2008.01.016
We have identified two genes in the genomic database for Caenorhabditis elegans that code for proteins with significant sequence similarity to the mammalian soluble epoxide hydrolase (sEH). The respective transcripts were cloned from a mixed stage cDNA library from C. elegans. The corresponding proteins obtained after recombinant expression in insect cells hydrolyzed standard epoxide hydrolase substrates, including epoxyeicosatrienoic acids (EETs) and leukotoxins (EpOMEs). The enzyme activity was inhibited by urea-based compounds originally designed to inhibit the mammalian sEH. In vivo inhibition of the enzymes using the most potent of these compounds resulted in elevated levels of the EpOMEs in the nematode. These results suggest that the hydrolases are involved in the metabolism of possible lipid signaling molecules in C. elegans.
Co-reporter:Jun-Yan Liu, Yan-Ping Lin, Hong Qiu, Christophe Morisseau, Tristan E. Rose, Sung Hee Hwang, Nipavan Chiamvimonvat, Bruce D. Hammock
European Journal of Pharmaceutical Sciences (12 March 2013) Volume 48(Issues 4–5) pp:619-627
Publication Date(Web):12 March 2013
DOI:10.1016/j.ejps.2012.12.013
Soluble epoxide hydrolase inhibitors (sEHIs) are anti-inflammatory, analgesic, anti-hypertensive, cardio- and renal-protective in multiple animal models. However, the earlier adamantyl-containing urea-based inhibitors are rapidly metabolized. Therefore, new potent inhibitors with the adamantyl group replaced by a substituted phenyl group were synthesized to presumptively offer better pharmacokinetic (PK) properties. Here we describe the improved PK profile of these inhibitors and the anti-inflammatory effect of the most promising one in a murine model. The PK profiles of inhibitors were determined following p.o. administration and serial bleeding in mice. The anti-inflammatory effect of 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl)urea (TPPU), the most promising inhibitor among the five sEHIs tested, was investigated in a lipopolysaccharide (LPS)-challenged murine model. The earlier broadly-used adamantyl-containing sEHI, trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-AUCB), was used for comparison. Compared with the earlier adamantyl-containing urea-based inhibitors, substituted phenyl-containing urea-based inhibitors afford more favorable PK properties, such as higher Cmaxs, larger AUCs and longer t1/2s, which, as expected, show more stable metabolic stability. Moreover, oral administration of TPPU dramatically reversed the shifts caused by LPS-challenge in plasma levels of inflammatory cytokines, epoxides and corresponding diols, which is more potent than t-AUCB. The substituted phenyl-containing sEHIs are more metabolically stable than those with adamantyl group, resulting in more potent efficacy in vivo. This indicates a new strategy for development of sEHIs for further study toward clinical trials.Download high-res image (106KB)Download full-size image
Co-reporter:Jun-Yan Liu, Hong Qiu, Christophe Morisseau, Sung Hee Hwang, Hsing-Ju Tsai, Arzu Ulu, Nipavan Chiamvimonvat, Bruce D. Hammock
Toxicology and Applied Pharmacology (1 September 2011) Volume 255(Issue 2) pp:200-206
Publication Date(Web):1 September 2011
DOI:10.1016/j.taap.2011.06.017
The increasing use of the antimicrobial triclocarban (TCC) in personal care products (PCPs) has resulted in concern regarding environmental pollution. TCC is a potent inhibitor of soluble epoxide hydrolase (sEH). Inhibitors of sEH (sEHIs) are anti-inflammatory, anti-hypertensive and cardio-protective in multiple animal models. However, the in vivo effects anticipated from a sEHI have not been reported for TCC. Here we demonstrated the anti-inflammatory effects in vivo of TCC in a murine model. TCC was employed in a lipopolysaccharide (LPS)-challenged murine model. Systolic blood pressure, plasma levels of several inflammatory cytokines and chemokine, and metabolomic profile of plasma oxylipins were determined. TCC significantly reversed LPS-induced morbid hypotension in a time-dependent manner. TCC significantly repressed the increased release of inflammatory cytokines and chemokine caused by LPS. Furthermore, TCC significantly shifted the oxylipin profile in vivo in a time-dependent manner towards resolution of inflammation as expected from a sEHI. These results demonstrated that at the doses used TCC is anti-inflammatory in the murine model. This study suggests that TCC may provide some benefits in humans in addition to its antimicrobial activities due to its potent inhibition of sEH. It may be a promising starting point for developing new low volume high value applications of TCC. However these biological effects also caution against the general over use of TCC in PCPs.Graphical abstractDownload high-res image (63KB)Download full-size imageResearch Highlights► Anti-microbial triclocarban (TCC) is anti-inflammatory in a murine model. ► TCC significantly shifted the oxylipin profile in vivo as expected from a sEHI. ► TCC significantly reversed LPS-induced morbid hypotension in a time-dependent manner. ► TCC significantly repressed LPS-induced increased release of inflammatory cytokines.
Co-reporter:Kin Sing Stephen Lee, Niel M. Henriksen, Connie J. Ng, Jun Yang, Weitao Jia, Christophe Morisseau, Armann Andaya, Michael K. Gilson, Bruce D. Hammock
Archives of Biochemistry and Biophysics (1 January 2017) Volume 613() pp:1-11
Publication Date(Web):1 January 2017
DOI:10.1016/j.abb.2016.10.017
•sEH inhibitor and EET photolabel mimics have been synthesized.•These photolabel mimics can be used to identify the binding site of EET in sEH.•The carboxylate of EET does not play an important role in its binding to sEH.•The binding orientation of EET is dictated by the stereochemistry of its epoxide.•A new computational model can predict how inhibitors and epoxides bind to sEH.Soluble epoxide hydrolase (sEH) is an important therapeutic target of many diseases, such as chronic obstructive pulmonary disease (COPD) and diabetic neuropathic pain. It acts by hydrolyzing and thus regulating specific bioactive long chain polyunsaturated fatty acid epoxides (lcPUFA), like epoxyeicosatrienoic acids (EETs). To better predict which epoxides could be hydrolyzed by sEH, one needs to dissect the important factors and structural requirements that govern the binding of the substrates to sEH. This knowledge allows further exploration of the physiological role played by sEH. Unfortunately, a crystal structure of sEH with a substrate bound has not yet been reported. In this report, new photoaffinity mimics of a sEH inhibitor and EET regioisomers were prepared and used in combination with peptide sequencing and computational modeling, to identify the binding orientation of different regioisomers and enantiomers of EETs into the catalytic cavity of sEH. Results indicate that the stereochemistry of the epoxide plays a crucial role in dictating the binding orientation of the substrate.Download high-res image (204KB)Download full-size image
Co-reporter:Sung Hee Hwang, Karen Wagner, Jian Xu, Jun Yang, Xichun Li, Zhengyu Cao, Christophe Morisseau, Kin Sing Stephen Lee, Bruce D. Hammock
Bioorganic & Medicinal Chemistry Letters (1 February 2017) Volume 27(Issue 3) pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.bmcl.2016.12.002
ω-Hydroxy polyunsaturated fatty acids (PUFAs), natural metabolites from arachidonic acid (ARA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) were prepared via convergent synthesis approach using two key steps: Cu-mediated CC bond formation to construct methylene skipped poly-ynes and a partial alkyne hydrogenation where the presence of excess 2-methyl-2-butene as an additive that is proven to be critical for the success of partial reduction of the poly-ynes to the corresponding cis-alkenes without over-hydrogenation. The potential biological function of ω-hydroxy PUFAs in pain was evaluated in naive rats. Following intraplantar injection, 20-hydroxyeicosatetraenoic acid (20-HETE, ω-hydroxy ARA) generated an acute decrease in paw withdrawal thresholds in a mechanical nociceptive assay indicating pain, but no change was observed from rats which received either 20-hydroxyeicosapentaenoic acid (20-HEPE, ω-hydroxy EPA) or 22-hydroxydocosahexaenoic acid (22-HDoHE, ω-hydroxy DHA). We also found that both 20-HEPE and 22-HDoHE are more potent than 20-HETE to activate murine transient receptor potential vanilloid receptor1 (mTRPV1).
Co-reporter:Stephen T. Vito, Adam T. Austin, Christopher N. Banks, Bora Inceoglu, Donald A. Bruun, Dorota Zolkowska, Daniel J. Tancredi, Michael A. Rogawski, Bruce D. Hammock, Pamela J. Lein
Toxicology and Applied Pharmacology (1 December 2014) Volume 281(Issue 2) pp:185-194
Publication Date(Web):1 December 2014
DOI:10.1016/j.taap.2014.10.001
•Acute TETS intoxication causes delayed and persistent neuroinflammation.•Diazepam given post-TETS prevents lethal tonic seizures but not neuroinflammation.•A soluble epoxide hydrolase inhibitor alters TETS-induced neuroinflammation.•Acute TETS intoxication may be more effectively treated by a combinatorial therapy.Tetramethylenedisulfotetramine (TETS) is a potent convulsant poison for which there is currently no approved antidote. The convulsant action of TETS is thought to be mediated by inhibition of type A gamma-aminobutyric acid receptor (GABAAR) function. We, therefore, investigated the effects of post-exposure administration of diazepam, a GABAAR positive allosteric modulator, on seizure activity, death and neuroinflammation in adult male Swiss mice injected with a lethal dose of TETS (0.15 mg/kg, ip). Administration of a high dose of diazepam (5 mg/kg, ip) immediately following the second clonic seizure (approximately 20 min post-TETS injection) effectively prevented progression to tonic seizures and death. However, this treatment did not prevent persistent reactive astrogliosis and microglial activation, as determined by GFAP and Iba-1 immunoreactivity and microglial cell morphology. Inhibition of soluble epoxide hydrolase (sEH) has been shown to exert potent anti-inflammatory effects and to increase survival in mice intoxicated with other GABAAR antagonists. The sEH inhibitor TUPS (1 mg/kg, ip) administered immediately after the second clonic seizure did not protect TETS-intoxicated animals from tonic seizures or death. Combined administration of diazepam (5 mg/kg, ip) and TUPS (1 mg/kg, ip, starting 1 h after diazepam and repeated every 24 h) prevented TETS-induced lethality and influenced signs of neuroinflammation in some brain regions. Significantly decreased microglial activation and enhanced reactive astrogliosis were observed in the hippocampus, with no changes in the cortex. Combining an agent that targets specific anti-inflammatory mechanisms with a traditional antiseizure drug may enhance treatment outcome in TETS intoxication.
Co-reporter:Sarunya Thiphom, Tippawan Prapamontol, Somporn Chantara, Ampica Mangklabruks, Chaisuree Suphavilai, Ki Chang Ahn, Shirley J. Gee and Bruce D. Hammock
Analytical Methods (2009-Present) 2012 - vol. 4(Issue 11) pp:NaN3778-3778
Publication Date(Web):2012/09/03
DOI:10.1039/C2AY25642H
The aim of this study was to identify a plasma biomarker of exposure to pyrethroid insecticides. A major metabolite, 3-phenoxybenzoic acid (3-PBA), can be detected in urine but urinary 3-PBA cannot be used to assess the active dose. The 3-PBA-adduct represents a much more persistent class of biomarkers than metabolites excreted into urine, having half-lives of up to several weeks or months. We developed an enzyme-linked immunosorbent assay (ELISA) for total 3-PBA including the adduct formed after alkaline hydrolysis, liquid–liquid extraction (LLE) and solid phase extraction (SPE) of the sample. The developed ELISA had an IC50 value of 26.7 ng mL−1. The intra- and inter-assay coefficients of variation (%CV) were lower than 5% and were within the optimum condition variance (OCV) range. The LLE cleanup technique satisfactorily eliminated the matrix effect from plasma samples before SPE and ELISA analysis yielding good recoveries (85.9–99.4%) with a limit of quantitation (LOQ, 5 ng mL−1) that was 30- to 47-fold more sensitive than previous studies. Moreover, the method developed could separate more than 80% of 3-PBA from the adducted form. The method was successfully applied for the detection of the target in real samples obtained from consumers (n = 50) and farmers (n = 50). To our knowledge, this is the first ELISA method for detecting 3-PBA in the human plasma that has been applied to a field study.
Co-reporter:Nils Helge Schebb, Bora Inceoglu, Tristan Rose, Karen Wagner and Bruce D. Hammock
Analytical Methods (2009-Present) 2011 - vol. 3(Issue 2) pp:
Publication Date(Web):
DOI:10.1039/C0AY00714E
Co-reporter:Bogdan Barnych, Amy A. Rand, Tomas Cajka, Kin Sing Stephen Lee and Bruce D. Hammock
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 20) pp:NaN4313-4313
Publication Date(Web):2017/04/25
DOI:10.1039/C7OB00789B
COX metabolites of 8,9-EET, previously observed as potent mitogenic lipid mediators, were synthesized for the first time by using two synthetic approaches. These synthetic materials allow for structural confirmation of COX metabolites of 8,9-EET and further study of their biological roles.

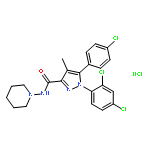
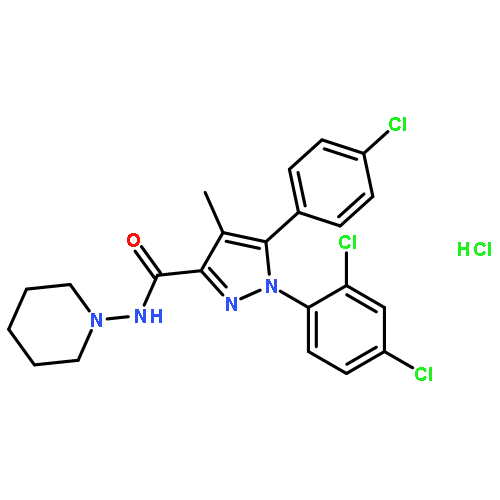

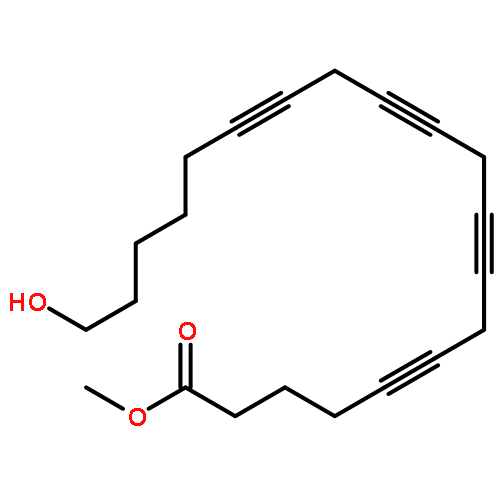

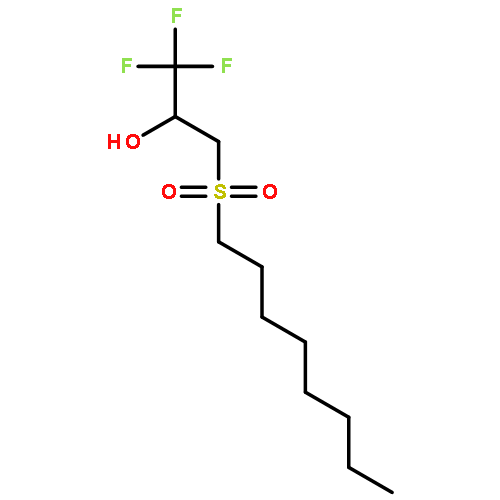
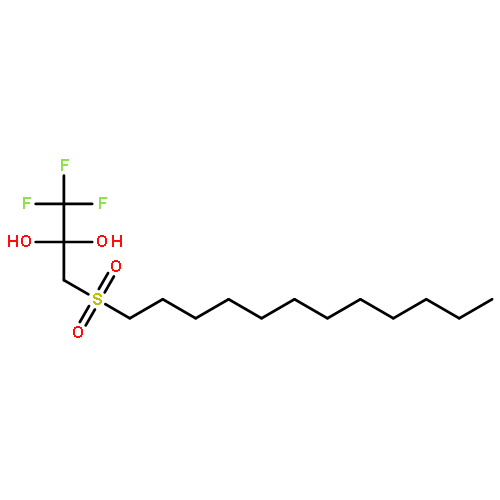
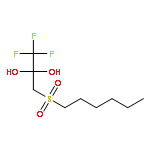
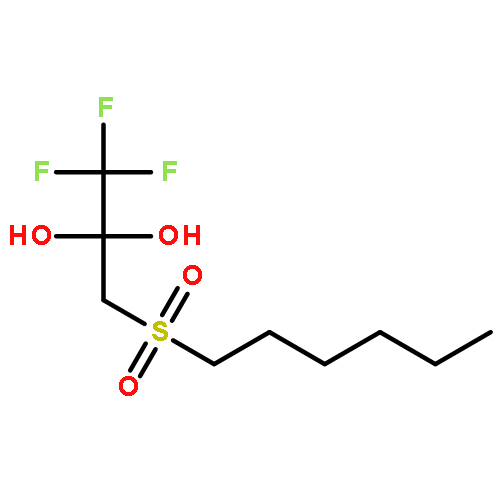
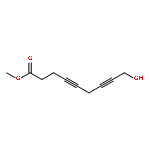
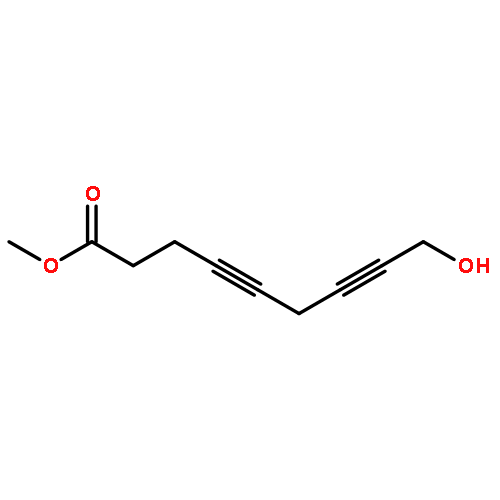
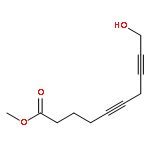
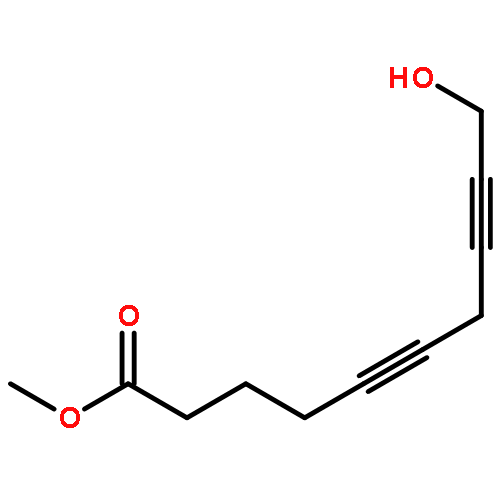




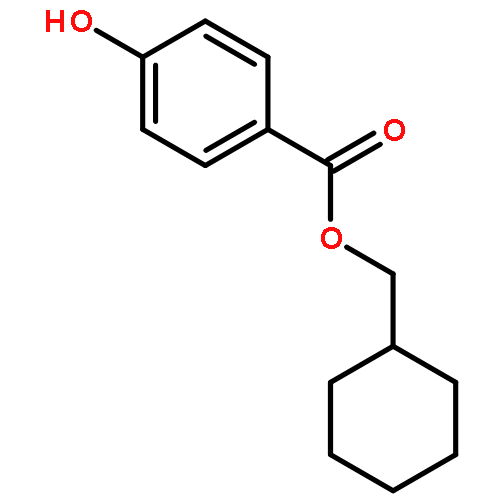
![2-Propanone, 1-[(2,6-dichlorophenyl)thio]-3,3,3-trifluoro-](http://img.cochemist.com/ccimg/111100/111045-29-5.png)
![2-Propanone, 1-[(2,6-dichlorophenyl)thio]-3,3,3-trifluoro-](http://img.cochemist.com/ccimg/111100/111045-29-5_b.png)
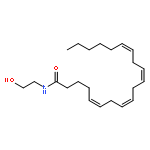
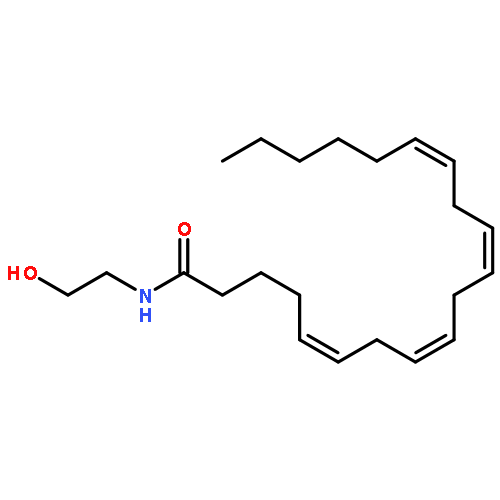
![2-Propanone, 3-[(4-chlorophenyl)thio]-1,1,1-trifluoro-](http://img.cochemist.com/ccimg/92700/92682-40-1.png)
![2-Propanone, 3-[(4-chlorophenyl)thio]-1,1,1-trifluoro-](http://img.cochemist.com/ccimg/92700/92682-40-1_b.png)
![2-Propanone, 1,1,1-trifluoro-3-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/92700/92682-36-5.png)
![2-Propanone, 1,1,1-trifluoro-3-[(4-methoxyphenyl)thio]-](http://img.cochemist.com/ccimg/92700/92682-36-5_b.png)
![2-Propanone, 1,1,1-trifluoro-3-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/92700/92682-32-1.png)
![2-Propanone, 1,1,1-trifluoro-3-[(4-methylphenyl)thio]-](http://img.cochemist.com/ccimg/92700/92682-32-1_b.png)
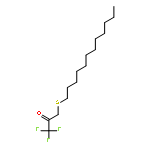
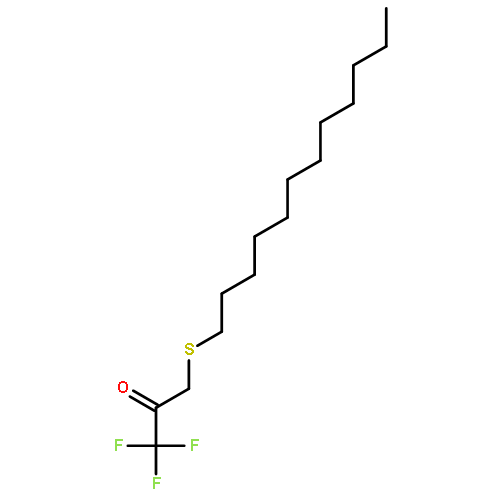

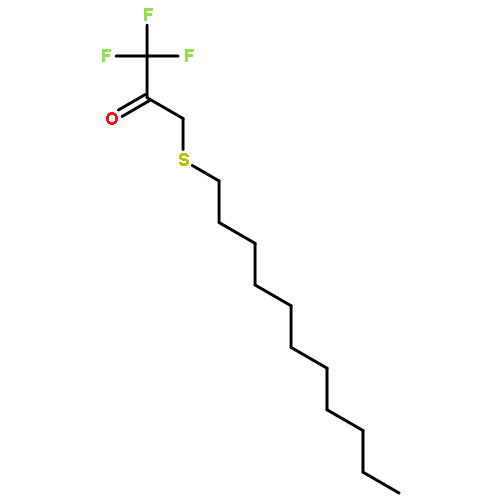

![2-Propanone, 1,1,1-trifluoro-3-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/90400/90357-44-1.png)
![2-Propanone, 1,1,1-trifluoro-3-[(4-nitrophenyl)thio]-](http://img.cochemist.com/ccimg/90400/90357-44-1_b.png)
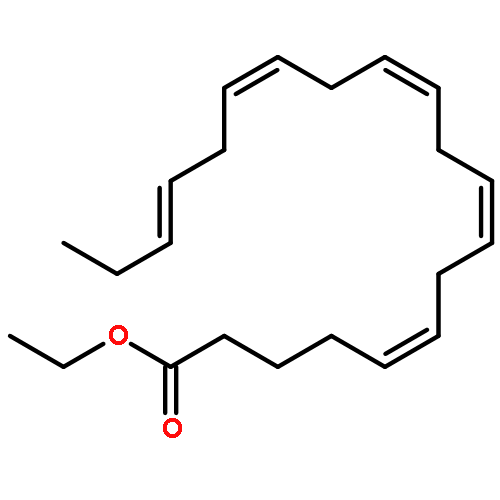










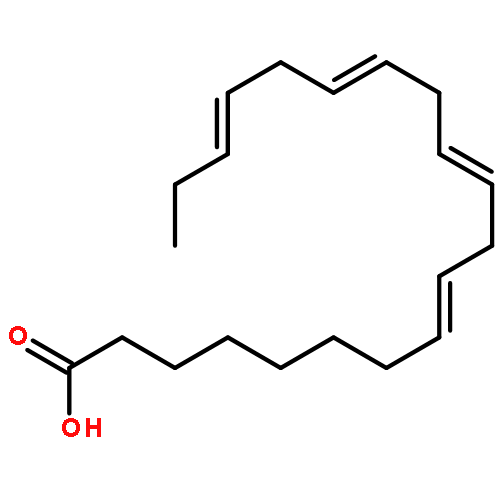






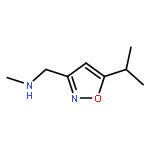
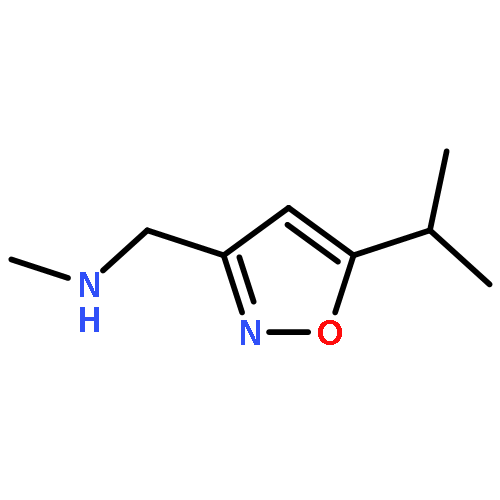
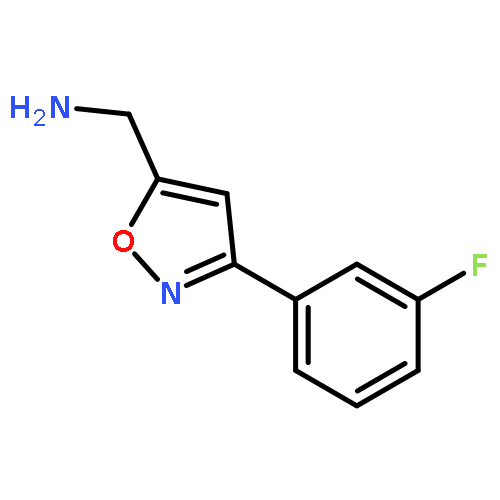
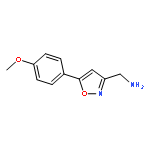
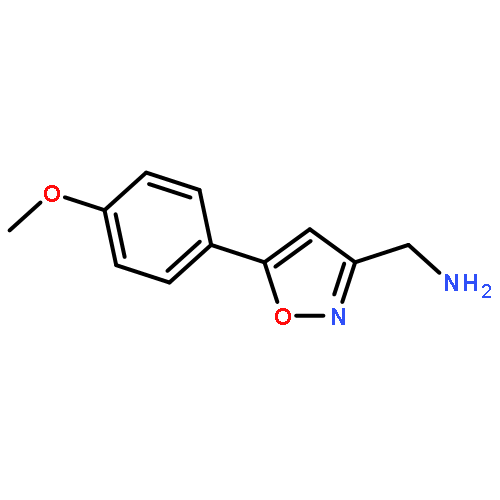
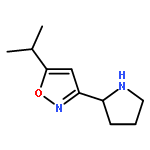
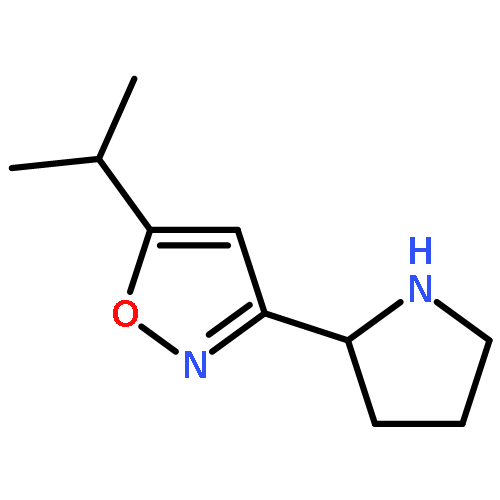

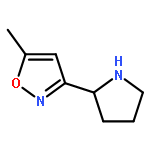
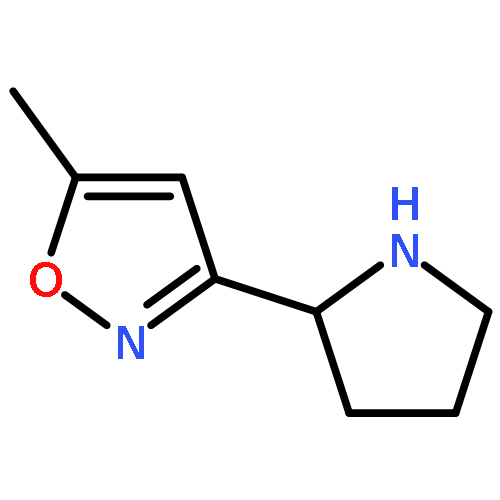
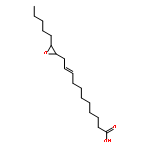
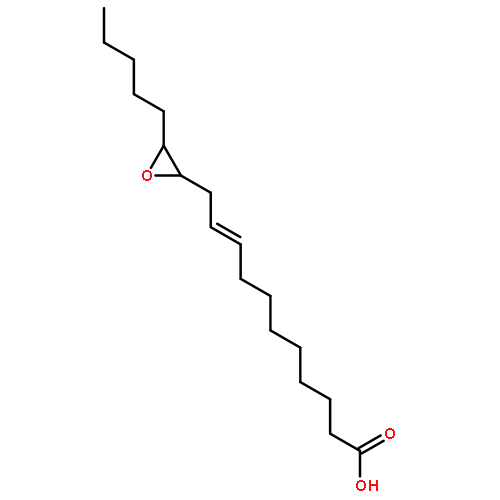



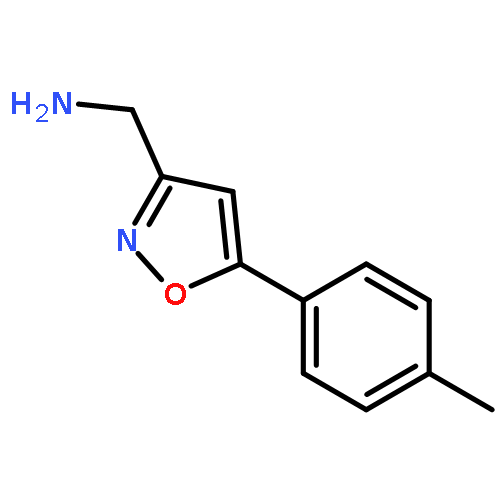

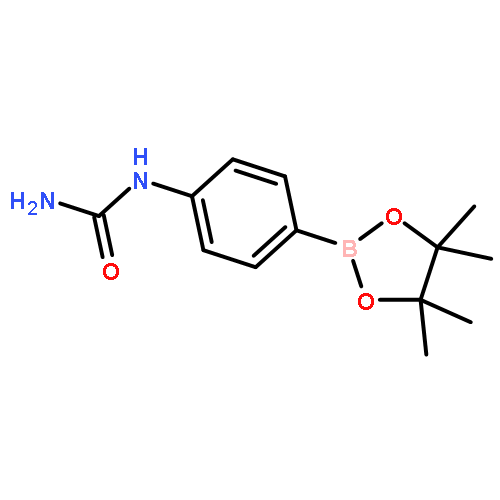
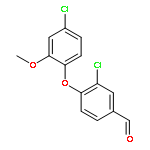
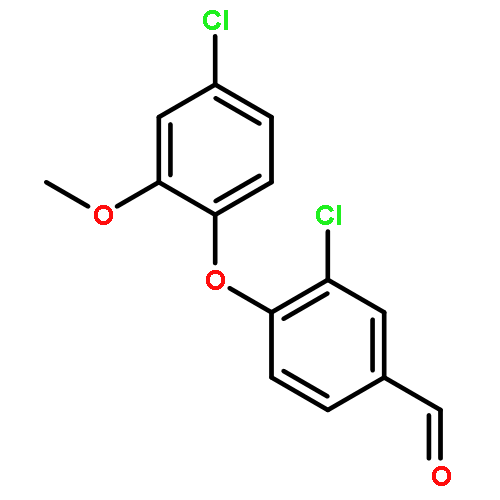
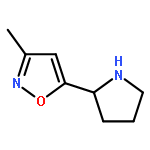
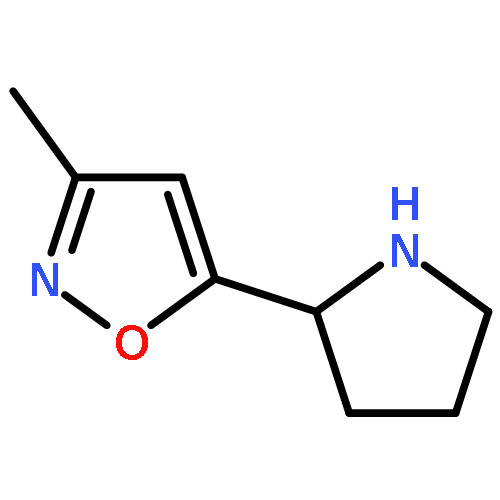
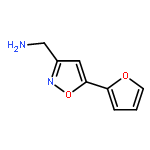



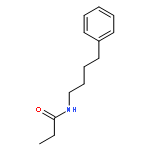
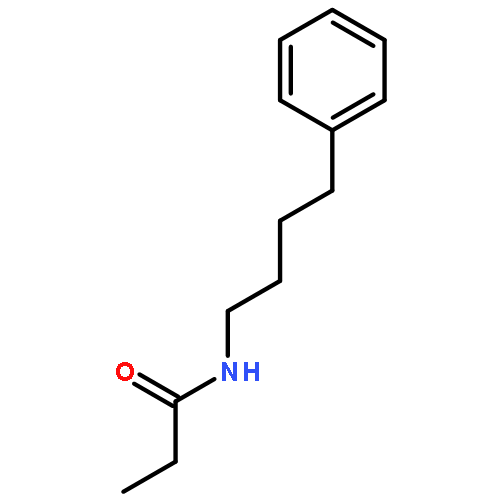


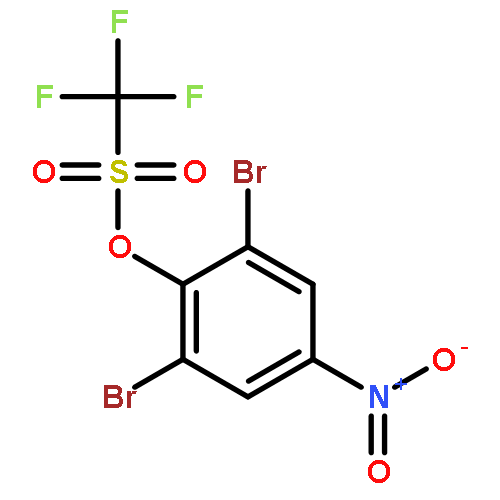
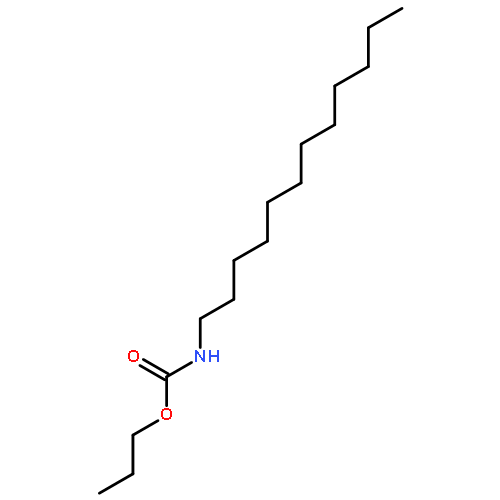
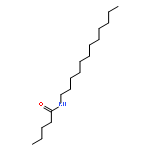
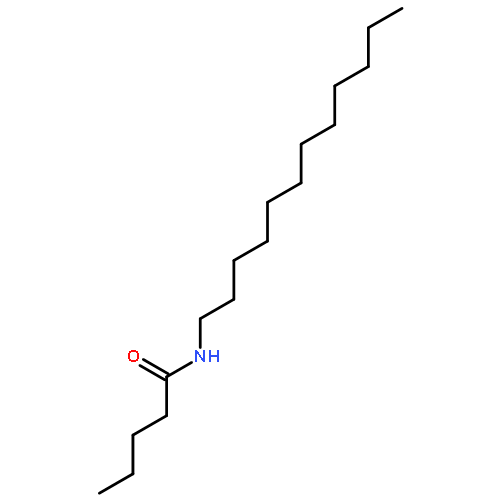
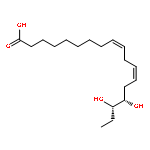
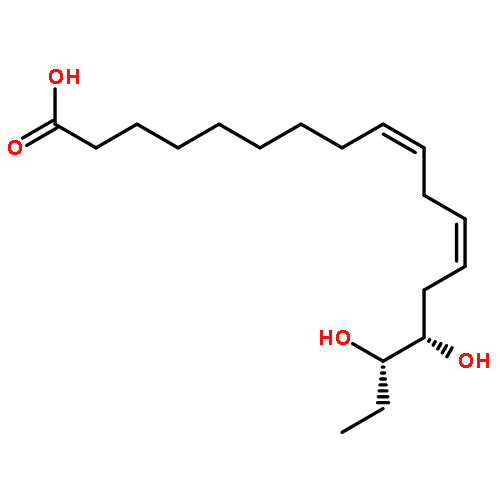


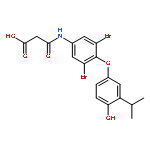
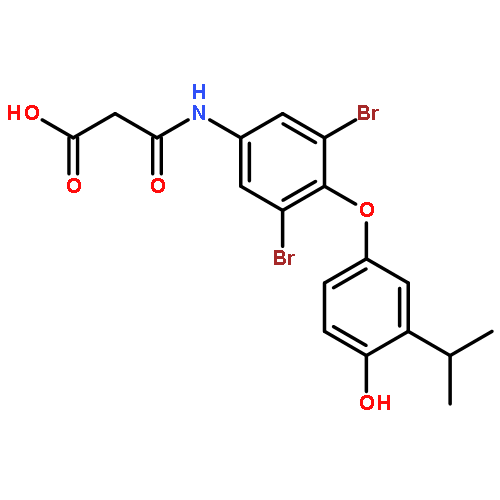










![Benzene, 1,3-dibromo-2-[4-methoxy-3-(1-methylethyl)phenoxy]-5-nitro-](http://img.cochemist.com/ccimg/258900/258820-25-6.png)
![Benzene, 1,3-dibromo-2-[4-methoxy-3-(1-methylethyl)phenoxy]-5-nitro-](http://img.cochemist.com/ccimg/258900/258820-25-6_b.png)


![Urea, N-[(4-methoxyphenyl)methyl]-N'-(phenylmethyl)-](http://img.cochemist.com/ccimg/189000/188911-54-8.png)
![Urea, N-[(4-methoxyphenyl)methyl]-N'-(phenylmethyl)-](http://img.cochemist.com/ccimg/189000/188911-54-8_b.png)
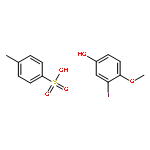















![Benzenamine, 3,5-dibromo-4-[4-methoxy-3-(1-methylethyl)phenoxy]-](http://img.cochemist.com/ccimg/169200/169113-83-1.png)
![Benzenamine, 3,5-dibromo-4-[4-methoxy-3-(1-methylethyl)phenoxy]-](http://img.cochemist.com/ccimg/169200/169113-83-1_b.png)
![L-Alaninamide,N-[(phenylmethoxy)carbonyl]-L-valyl-N-[(1S)-1-(carboxymethyl)-3-fluoro-2-oxopropyl]-](http://img.cochemist.com/ccimg/161500/161401-82-7.png)
![L-Alaninamide,N-[(phenylmethoxy)carbonyl]-L-valyl-N-[(1S)-1-(carboxymethyl)-3-fluoro-2-oxopropyl]-](http://img.cochemist.com/ccimg/161500/161401-82-7_b.png)


![Urea, N-4-piperidinyl-N'-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/160500/160431-85-6.png)
![Urea, N-4-piperidinyl-N'-[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/160500/160431-85-6_b.png)
![Benzene, 1,3-dichloro-2-[4-methoxy-3-(1-methylethyl)phenoxy]-5-nitro-](http://img.cochemist.com/ccimg/156800/156740-82-8.png)
![Benzene, 1,3-dichloro-2-[4-methoxy-3-(1-methylethyl)phenoxy]-5-nitro-](http://img.cochemist.com/ccimg/156800/156740-82-8_b.png)



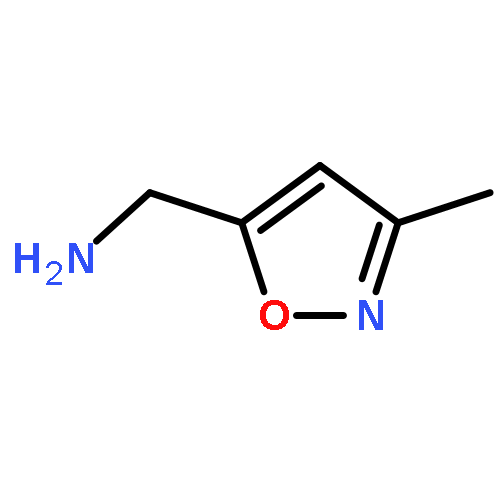









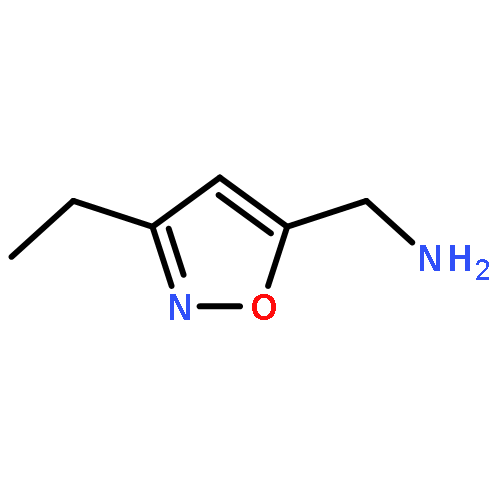










![5,8,11-Tridecatrienoicacid, 13-[(2R,3S)-3-(2Z)-2-penten-1-yl-2-oxiranyl]-, (5Z,8Z,11Z)-rel-](http://img.cochemist.com/ccimg/131400/131339-24-7.png)
![5,8,11-Tridecatrienoicacid, 13-[(2R,3S)-3-(2Z)-2-penten-1-yl-2-oxiranyl]-, (5Z,8Z,11Z)-rel-](http://img.cochemist.com/ccimg/131400/131339-24-7_b.png)
![5,8,11,14-Hexadecatetraenoicacid, 16-[(2R,3S)-3-ethyl-2-oxiranyl]-, (5Z,8Z,11Z,14Z)-rel-](http://img.cochemist.com/ccimg/131400/131339-23-6.png)
![5,8,11,14-Hexadecatetraenoicacid, 16-[(2R,3S)-3-ethyl-2-oxiranyl]-, (5Z,8Z,11Z,14Z)-rel-](http://img.cochemist.com/ccimg/131400/131339-23-6_b.png)














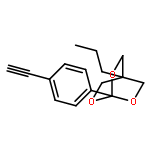
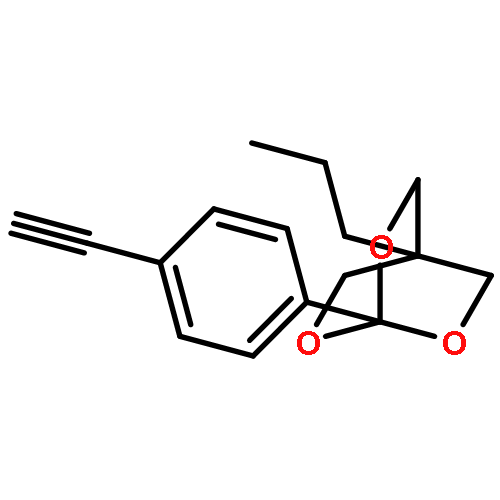






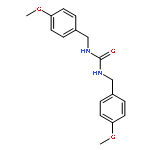

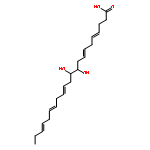

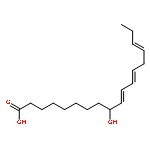
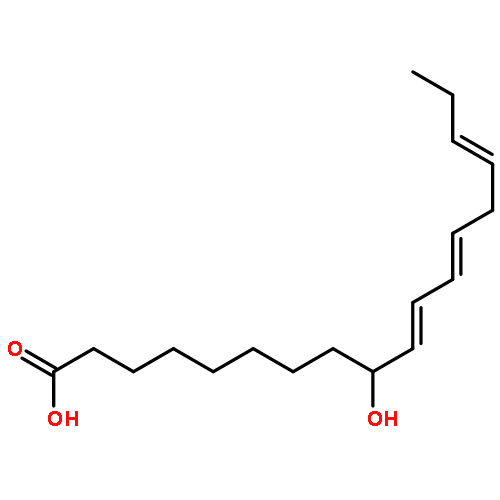
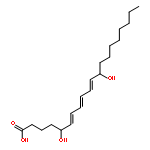




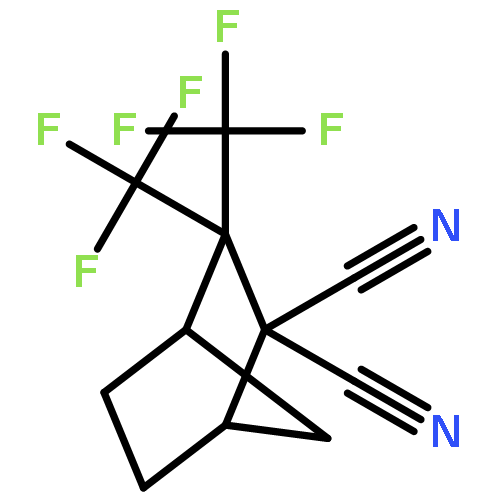
![(5Z)-7-{(2R,3S)-3-[(2Z,5E)-undeca-2,5-dien-1-yl]oxiran-2-yl}hept-5-enoic acid](http://img.cochemist.com/ccimg/82900/82864-43-5.png)
![(5Z)-7-{(2R,3S)-3-[(2Z,5E)-undeca-2,5-dien-1-yl]oxiran-2-yl}hept-5-enoic acid](http://img.cochemist.com/ccimg/82900/82864-43-5_b.png)
![5-Heptenoic acid,7-[3-(2,5-undecadien-1-yl)-2-oxiranyl]-](http://img.cochemist.com/ccimg/81300/81246-85-7.png)
![5-Heptenoic acid,7-[3-(2,5-undecadien-1-yl)-2-oxiranyl]-](http://img.cochemist.com/ccimg/81300/81246-85-7_b.png)

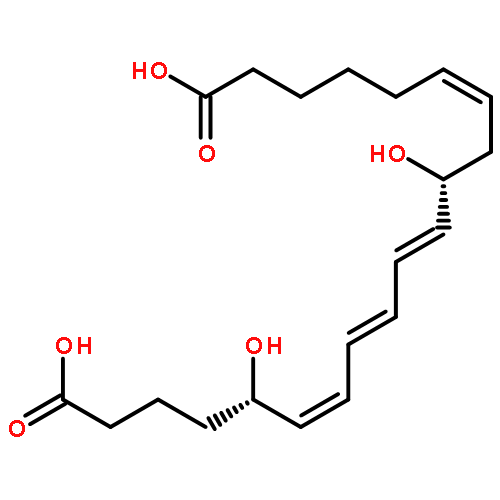




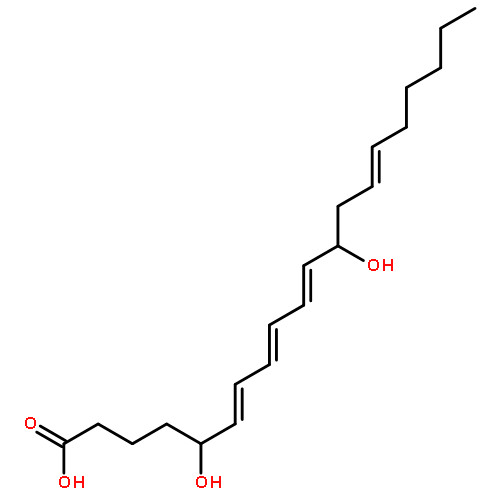
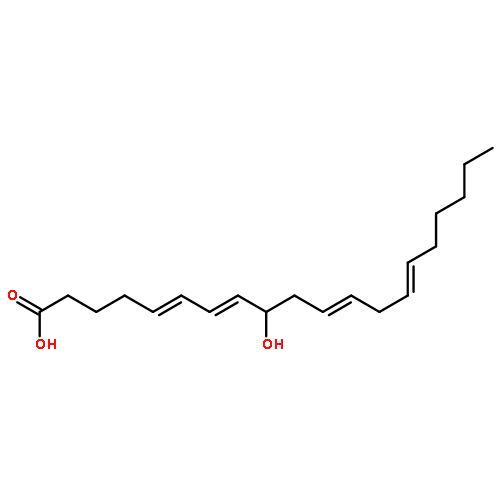
![TRICYCLO[3.3.1.13,7]DECANE, 1-(ISOCYANATOMETHYL)-](http://img.cochemist.com/ccimg/69900/69887-12-3.png)
![TRICYCLO[3.3.1.13,7]DECANE, 1-(ISOCYANATOMETHYL)-](http://img.cochemist.com/ccimg/69900/69887-12-3_b.png)

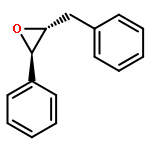
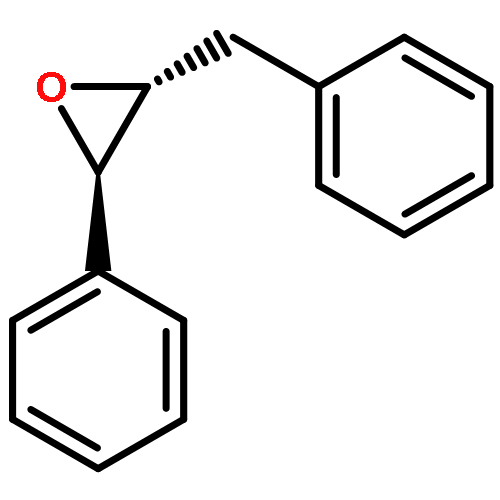


![(9Z,12Z)-14-[(2R,3S)-3-ethyloxiran-2-yl]tetradeca-9,12-dienoic acid](http://img.cochemist.com/ccimg/64100/64044-08-2.png)
![(9Z,12Z)-14-[(2R,3S)-3-ethyloxiran-2-yl]tetradeca-9,12-dienoic acid](http://img.cochemist.com/ccimg/64100/64044-08-2_b.png)


![8-{(2R)-2-[(3E)-oct-3-en-1-yl]oxiran-2-yl}octanoic acid](http://img.cochemist.com/ccimg/62000/61949-82-4.png)
![8-{(2R)-2-[(3E)-oct-3-en-1-yl]oxiran-2-yl}octanoic acid](http://img.cochemist.com/ccimg/62000/61949-82-4_b.png)
![2,6,7-Trioxa-1-phosphabicyclo[2.2.2]octane,4-(1,1-dimethylethyl)-, 1-oxide](http://img.cochemist.com/ccimg/61500/61481-19-4.png)
![2,6,7-Trioxa-1-phosphabicyclo[2.2.2]octane,4-(1,1-dimethylethyl)-, 1-oxide](http://img.cochemist.com/ccimg/61500/61481-19-4_b.png)


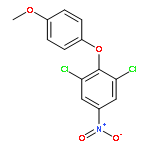
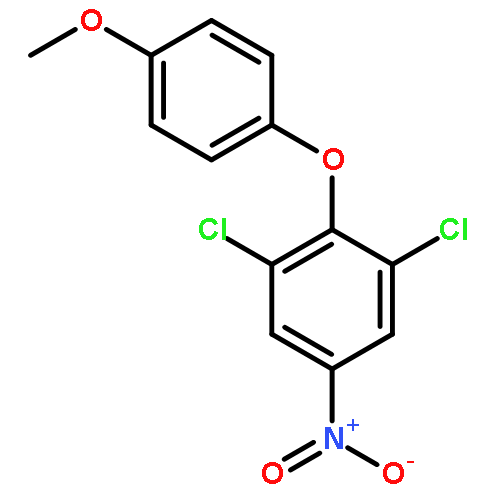


![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0.png)
![7-[3-(3-HYDROXYOCT-1-ENYL)-4,6-DIOXABICYCLO[3.1.1]HEPT-2-YL]HEPT-5-ENOIC ACID](http://img.cochemist.com/ccimg/57600/57576-52-0_b.png)
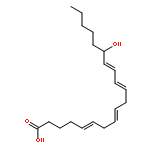


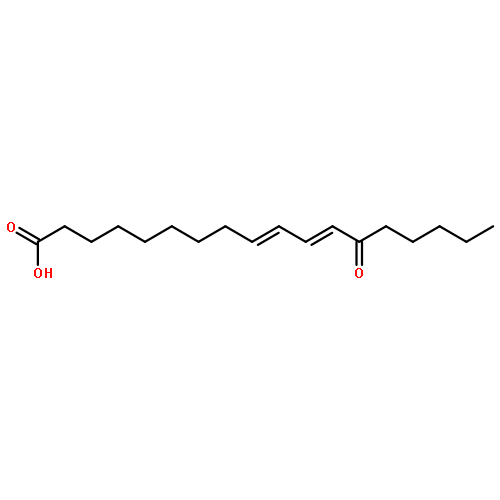
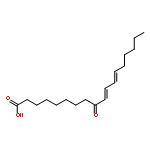
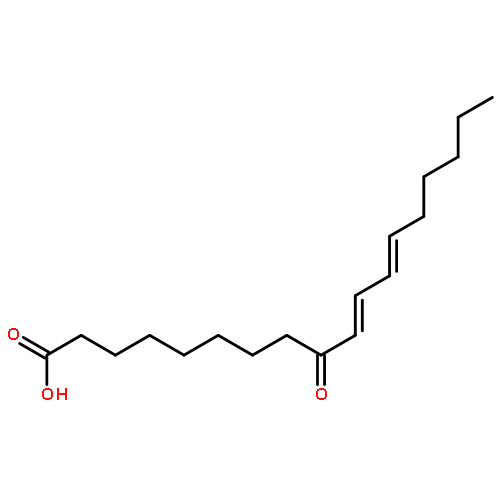







![9-THIA-1,3,6,8-TETRAAZATRICYCLO[4.3.1.1(3,8)]UNDECANE, 9,9-DIOXIDE](http://img.cochemist.com/ccimg/37400/37391-75-6.png)
![9-THIA-1,3,6,8-TETRAAZATRICYCLO[4.3.1.1(3,8)]UNDECANE, 9,9-DIOXIDE](http://img.cochemist.com/ccimg/37400/37391-75-6_b.png)
![8-[(2R)-2-(octa-5,6-dien-4-yl)oxiran-2-yl]octanoic acid](http://img.cochemist.com/ccimg/36800/36747-82-7.png)
![8-[(2R)-2-(octa-5,6-dien-4-yl)oxiran-2-yl]octanoic acid](http://img.cochemist.com/ccimg/36800/36747-82-7_b.png)






![2,7:3,6-Dimethanonaphth[2,3-b]oxiren-8-one,3,4,5,6,9,9-hexachloro-1a,2,2a,3,6,6a,7,7a-octahydro-,(1aR,2R,2aR,3R,6S,6aS,7S,7aS)-rel- (9CI)](http://img.cochemist.com/ccimg/28600/28548-08-5.png)
![2,7:3,6-Dimethanonaphth[2,3-b]oxiren-8-one,3,4,5,6,9,9-hexachloro-1a,2,2a,3,6,6a,7,7a-octahydro-,(1aR,2R,2aR,3R,6S,6aS,7S,7aS)-rel- (9CI)](http://img.cochemist.com/ccimg/28600/28548-08-5_b.png)
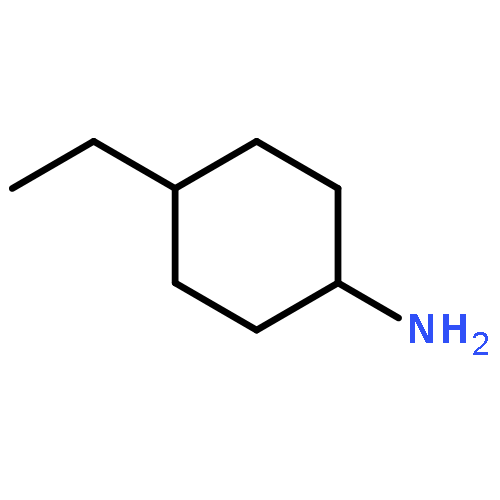
![2,6-Nonadienoic acid,9-[(2R)-3,3-dimethyl-2-oxiranyl]-3,7-dimethyl-, methyl ester, (2E,6E)-](http://img.cochemist.com/ccimg/23000/22963-93-5.png)
![2,6-Nonadienoic acid,9-[(2R)-3,3-dimethyl-2-oxiranyl]-3,7-dimethyl-, methyl ester, (2E,6E)-](http://img.cochemist.com/ccimg/23000/22963-93-5_b.png)
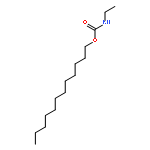
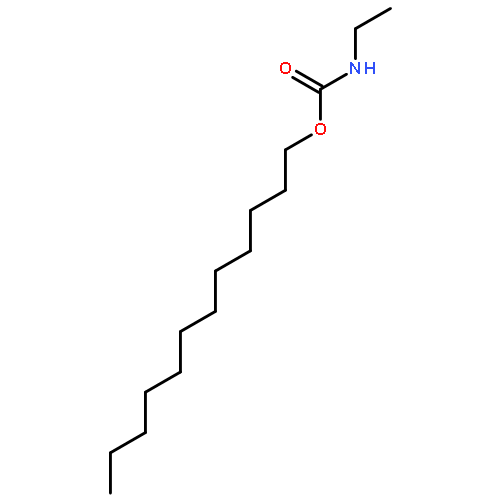




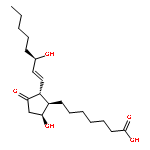
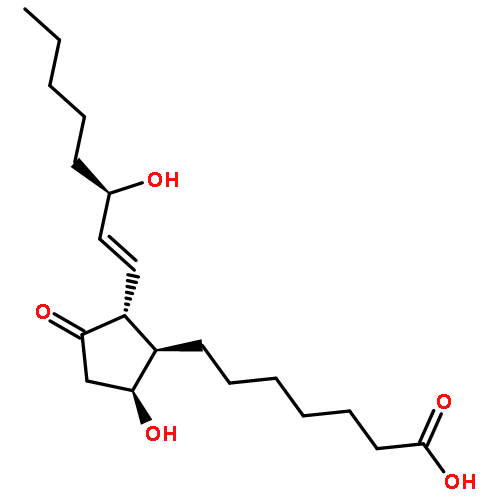
![3,6-Methano-8H-1,5,7-trioxacyclopenta[ij]cycloprop[a]azulene-4,8(3H)-dione,hexahydro-2a-hydroxy-8b-methyl-9-(1-methylethenyl)-,(1aR,2aR,3S,6R,6aS,8aS,8bR,9R)-](http://img.cochemist.com/ccimg/17700/17617-45-7.png)
![3,6-Methano-8H-1,5,7-trioxacyclopenta[ij]cycloprop[a]azulene-4,8(3H)-dione,hexahydro-2a-hydroxy-8b-methyl-9-(1-methylethenyl)-,(1aR,2aR,3S,6R,6aS,8aS,8bR,9R)-](http://img.cochemist.com/ccimg/17700/17617-45-7_b.png)



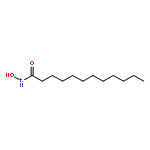


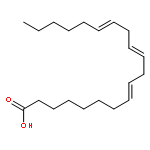












![Benzenamine, 4-[1,2,2,2-tetrafluoro-1-(trifluoromethyl)ethyl]-](http://img.cochemist.com/ccimg/2400/2396-17-0.png)
![Benzenamine, 4-[1,2,2,2-tetrafluoro-1-(trifluoromethyl)ethyl]-](http://img.cochemist.com/ccimg/2400/2396-17-0_b.png)

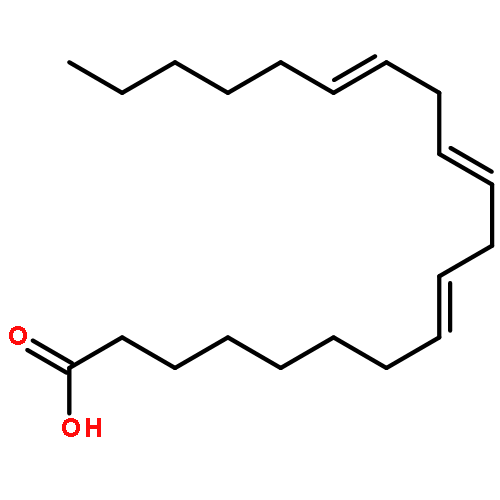












![(2R)-2-AMINO-1-PHENYL-PROPAN-1-OL; (E)-4-(4-BROMOPHENOXY)-4-OXO-B<WBR />UT-2-ENOIC ACID; N,N-DIMETHYL-3-(2-PYRIDYL)PROPAN-1-AMINE; 3-[(1R<WBR />)-1-HYDROXY-2-METHYLAMINO-ETHYL]PHENOL; DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/400/320-77-4.png)
![(2R)-2-AMINO-1-PHENYL-PROPAN-1-OL; (E)-4-(4-BROMOPHENOXY)-4-OXO-B<WBR />UT-2-ENOIC ACID; N,N-DIMETHYL-3-(2-PYRIDYL)PROPAN-1-AMINE; 3-[(1R<WBR />)-1-HYDROXY-2-METHYLAMINO-ETHYL]PHENOL; DIHYDROCHLORIDE](http://img.cochemist.com/ccimg/400/320-77-4_b.png)
![2,8,9-Trioxa-5-aza-1-silabicyclo[3.3.3]undecane](http://img.cochemist.com/ccimg/300/283-60-3.png)
![2,8,9-Trioxa-5-aza-1-silabicyclo[3.3.3]undecane](http://img.cochemist.com/ccimg/300/283-60-3_b.png)



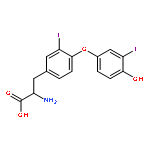

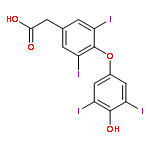
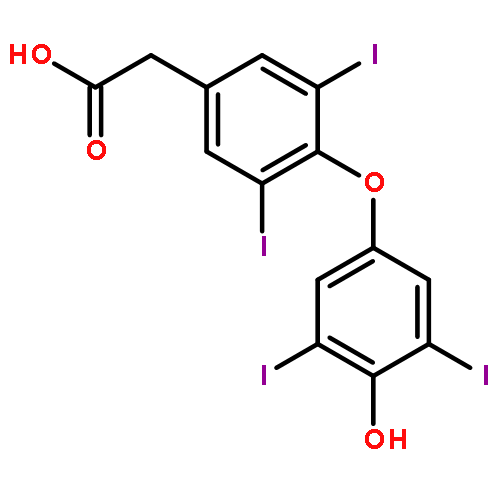








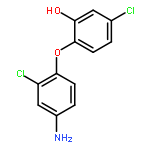
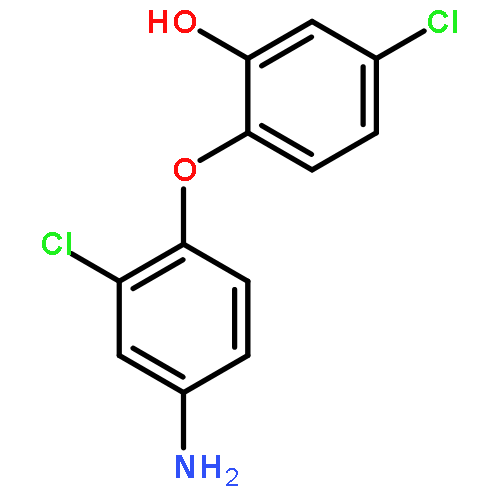
![8,11,14-Trioxa-2-azahexadecanamide, N-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/776400/776300-69-7.png)
![8,11,14-Trioxa-2-azahexadecanamide, N-tricyclo[3.3.1.13,7]dec-1-yl-](http://img.cochemist.com/ccimg/776400/776300-69-7_b.png)















![(5Z,8Z)-10-{(2R,3S)-3-[(2Z)-oct-2-en-1-yl]oxiran-2-yl}deca-5,8-dienoic acid](http://img.cochemist.com/ccimg/87200/87173-81-7.png)
![(5Z,8Z)-10-{(2R,3S)-3-[(2Z)-oct-2-en-1-yl]oxiran-2-yl}deca-5,8-dienoic acid](http://img.cochemist.com/ccimg/87200/87173-81-7_b.png)




![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)