Co-reporter:Jeffrey E. Lopez, Sarah E. Haynes, Jaimeen D. Majmudar, Brent R. Martin, and Carol A. Fierke
Journal of the American Chemical Society November 15, 2017 Volume 139(Issue 45) pp:16222-16222
Publication Date(Web):October 16, 2017
DOI:10.1021/jacs.7b07603
The histone deacetylase family comprises 18 enzymes that catalyze deacetylation of acetylated lysine residues; however, the specificity and substrate profile of each isozyme remains largely unknown. Due to transient enzyme–substrate interactions, conventional co-immunoprecipitation methods frequently fail to identify enzyme-specific substrates. Additionally, compensatory mechanisms often limit the ability of knockdown or chemical inhibition studies to achieve significant fold changes observed by acetylation proteomics methods. Furthermore, measured alterations do not guarantee a direct link between enzyme and substrate. Here we present a chemical crosslinking strategy that incorporates a photoreactive, non-natural amino acid, p-benzoyl-l-phenylalanine, into various positions of the structurally characterized isozyme histone deacetylase 8 (HDAC8). After covalent capture, co-immunoprecipitation, and mass spectrometric analysis, we identified a subset of HDAC8 substrates from human cell lysates, which were further validated for catalytic turnover. Overall, this chemical crosslinking approach identified novel HDAC8-specific substrates with high catalytic efficiency, thus presenting a general strategy for unbiased deacetylase substrate discovery.
Co-reporter:Sarah E. Haynes, Daniel A. Polasky, Sugyan M. Dixit, Jaimeen D. Majmudar, Kieran Neeson, Brandon T. Ruotolo, and Brent R. Martin
Analytical Chemistry June 6, 2017 Volume 89(Issue 11) pp:5669-5669
Publication Date(Web):May 4, 2017
DOI:10.1021/acs.analchem.7b00112
High mass accuracy, data-dependent acquisition is the current standard method in mass spectrometry-based peptide annotation and quantification. In high complexity samples, limited instrument scan speeds often result in under-sampling. In contrast, all-ion data-independent acquisition methods bypass precursor selection, alternating high and low collision energies to analyze product and precursor ions across wide mass ranges. Despite capturing data for all events, peptide annotation is limited by inadequate alignment algorithms or overlapping ions. Ion mobility separation can add an orthogonal analytical dimension, reducing ion interference to improve reproducibility, peak capacity, and peptide identifications to rival modern hybrid quadrupole orbitrap systems. Despite the advantages of ion mobility separation in complex proteomics analyses, there has been no quantitative measure of ion mobility resolution in a complex proteomic sample. Here, we present TWIMExtract, a data extraction tool to export defined slices of liquid chromatography/ion mobility/mass spectrometry (LC-IM-MS) data, providing a route to quantify ion mobility resolution from a commercial traveling-wave ion mobility time-of-flight mass spectrometer. Using standard traveling-wave ion mobility parameters (600 m/s, 40 V), 90% of the annotated peptides occupied just 23% of the ion mobility drift space, yet inclusion of ion mobility nearly doubled the overall peak capacity. Relative to fixed velocity traveling-wave ion mobility settings, ramping the traveling-wave velocity increased drift space occupancy, amplifying resolution by 16%, peak capacity by nearly 50%, and peptide/protein identifications by 40%. Overall, variable-velocity traveling-wave ion mobility-mass spectrometry significantly enhances proteomics analysis in all-ion fragmentation acquisition.
Co-reporter:Sang Joon Won;Dahvid Davda;Sin Ye Hwang;Kristin J. Labby;Rachel Pricer;Kira A. Armacost;Jaimeen D. Majmudar;Fei San Chong;Edward S. Hur;Laura A. Rodriguez;Jasmine Palakurthi;Jennifer L. Meagher;Brent R. Martin;Christina L. Rodriguez;Charles L. Brooks;Jeanne A. Stuckey;Kristopher A. Torossian
ACS Chemical Biology December 16, 2016 Volume 11(Issue 12) pp:3374-3382
Publication Date(Web):October 17, 2016
DOI:10.1021/acschembio.6b00720
Post-translational S-palmitoylation directs the trafficking and membrane localization of hundreds of cellular proteins, often involving a coordinated palmitoylation cycle that requires both protein acyl transferases (PATs) and acyl protein thioesterases (APTs) to actively redistribute S-palmitoylated proteins toward different cellular membrane compartments. This process is necessary for the trafficking and oncogenic signaling of S-palmitoylated Ras isoforms, and potentially many peripheral membrane proteins. The depalmitoylating enzymes APT1 and APT2 are separately conserved in all vertebrates, suggesting unique functional roles for each enzyme. The recent discovery of the APT isoform-selective inhibitors ML348 and ML349 has opened new possibilities to probe the function of each enzyme, yet it remains unclear how each inhibitor achieves orthogonal inhibition. Herein, we report the high-resolution structure of human APT2 in complex with ML349 (1.64 Å), as well as the complementary structure of human APT1 bound to ML348 (1.55 Å). Although the overall peptide backbone structures are nearly identical, each inhibitor adopts a distinct conformation within each active site. In APT1, the trifluoromethyl group of ML348 is positioned above the catalytic triad, but in APT2, the sulfonyl group of ML349 forms hydrogen bonds with active site resident waters to indirectly engage the catalytic triad and oxyanion hole. Reciprocal mutagenesis and activity profiling revealed several differing residues surrounding the active site that serve as critical gatekeepers for isoform accessibility and dynamics. Structural and biochemical analysis suggests the inhibitors occupy a putative acyl-binding region, establishing the mechanism for isoform-specific inhibition, hydrolysis of acyl substrates, and structural orthogonality important for future probe development.
Co-reporter:Nicholas B. Borotto, Phillip J. McClory, Brent R. Martin, and Kristina Håkansson
Analytical Chemistry August 15, 2017 Volume 89(Issue 16) pp:8304-8304
Publication Date(Web):July 14, 2017
DOI:10.1021/acs.analchem.7b01461
Protein S-sulfinylation (R–SO2–) and S-sulfonylation (R–SO3–) are irreversible oxidative post-translational modifications of cysteine residues. Greater than 5% of cysteines are reported to occupy these higher oxidation states, which effectively inactivate the corresponding thiols and alter the electronic and physical properties of modified proteins. Such higher oxidation states are reached after excessive exposure to cellular oxidants, and accumulate across different disease states. Despite widespread and functionally relevant cysteine oxidation across the proteome, there are currently no robust methods to profile higher order cysteine oxidation. Traditional data-dependent liquid chromatography/tandem mass spectrometry (LC/MS/MS) methods generally miss low-occupancy modifications in complex analyses. Here, we present a data-independent acquisition (DIA) LC/MS-based approach, leveraging the high IR absorbance of sulfoxides at 10.6 μm, for selective dissociation and discovery of S-sulfonated peptides. Across peptide standards and protein digests, we demonstrate selective infrared multiphoton dissociation (IRMPD) of S-sulfonated peptides in the background of unmodified peptides. This selective DIA IRMPD LC/MS-based approach allows identification and annotation of S-sulfonated peptides across complex mixtures while providing sufficient sequence information to localize the modification site.
Co-reporter:Christopher T. M. B. Tom;John E. Crellin;Hashim F. Motiwala;Matthew B. Stone;Dahvid Davda;William Walker;Yu-Hsuan Kuo;Jeannie L. Hernandez;Kristin J. Labby;Lyanne Gomez-Rodriguez;Paul M. Jenkins;Sarah L. Veatch;Brent R. Martin
Chemical Communications 2017 vol. 53(Issue 53) pp:7385-7388
Publication Date(Web):2017/06/29
DOI:10.1039/C7CC02285A
Here we report a ratiometric fluorescent probe for chemoselective conjugation to sulfenic acids in living cells. Our approach couples an α-fluoro-substituted dimedone to an aminonaphthalene fluorophore (F-DiNap), which upon sulfenic acid conjugation is locked as the 1,3-diketone, changing the fluorophore excitation. F-DiNap reacts with S-sulfenylated proteins at equivalent rates to current probes, but the α-fluorine substitution blocks side-reactions with biological aldehydes.
Co-reporter:Sang Joon Won, Joseph D. Eschweiler, Jaimeen D. Majmudar, Fei San Chong, Sin Ye Hwang, Brandon T. Ruotolo, and Brent R. Martin
ACS Medicinal Chemistry Letters 2017 Volume 8(Issue 2) pp:
Publication Date(Web):December 9, 2016
DOI:10.1021/acsmedchemlett.6b00441
Activity-based protein profiling (ABPP) has revolutionized the discovery and optimization of active-site ligands across distinct enzyme families, providing a robust platform for in-class selectivity profiling. Nonetheless, this approach is less straightforward for profiling reversible inhibitors and does not access proteins outside the ABPP probe’s target profile. While the active-site competitive acyl protein thioesterase 2 inhibitor ML349 (Ki = 120 nM) is highly selective within the serine hydrolase enzyme family, it could still interact with other cellular targets. Here we present a chemoproteomic workflow to enrich and profile candidate ML349-binding proteins. In human cell lysates, biotinylated-ML349 enriches a recurring set of proteins, including metabolite kinases and flavin-dependent oxidoreductases that are potentially enhanced by avidity-driven multimeric interactions. Confirmatory assays by native mass spectrometry and fluorescence polarization quickly rank-ordered these weak off-targets, providing justification to explore ligand interactions and stoichiometry beyond ABPP.Keywords: Activity-based protein profiling; chemical proteomics; inhibitor selectivity; native mass spectrometry;
Co-reporter:Jeannie L. Hernandez, Dahvid Davda, Melanie Cheung See Kit, Jaimeen D. Majmudar, ... Brent R. Martin
Cell Chemical Biology 2017 Volume 24, Issue 1(Volume 24, Issue 1) pp:
Publication Date(Web):19 January 2017
DOI:10.1016/j.chembiol.2016.12.007
•Scribble S-palmitoylation is reduced after overexpression of Snail•Snail overexpression reduces levels of select zDHHC protein acyl transferases•After Snail overexpression, APT2 inhibition rescues Scribble localization•MEK activation is attenuated following APT2 inhibition in Snail-expressing cellsThe multidomain scaffolding protein Scribble (Scrib) organizes key signaling complexes to specify basolateral cell polarity and suppress aberrant growth. In many human cancers, genetically normal Scrib mislocalizes from cell-cell junctions to the cytosol, correlating with enhanced growth signaling and malignancy. Here we confirm that expression of the epithelial-to-mesenchymal transcription factor (EMT-TF) Snail in benign epithelial cells leads to Scrib displacement from the plasma membrane, mimicking the mislocalization observed in aggressive cancers. Upon further examination, Snail promotes a transcriptional program that targets genes in the palmitoylation cycle, repressing many protein acyl transferases and elevating expression and activity of protein acyl thioesterase 2 (APT2). APT2 isoform-selective inhibition or knockdown rescued Scrib membrane localization and palmitoylation while attenuating MEK activation. Overall, inhibiting APT2 restores balance to the Scrib palmitoylation cycle, promoting membrane re-localization and growth attenuation. These findings emphasize the importance of S-palmitoylation as a post-translational gatekeeper of cell polarity-mediated tumor suppression.Download high-res image (438KB)Download full-size image
Co-reporter:Jaimeen D. Majmudar; Aaron M. Konopko; Kristin J. Labby; Christopher T. M. B. Tom; John E. Crellin; Ashesh Prakash;Brent R. Martin
Journal of the American Chemical Society 2016 Volume 138(Issue 6) pp:1852-1859
Publication Date(Web):January 19, 2016
DOI:10.1021/jacs.5b06806
Cysteine S-nitrosation and S-sulfination are naturally occurring post-translational modifications (PTMs) on proteins induced by physiological signals and redox stress. Here we demonstrate that sulfinic acids and nitrosothiols react to form a stable thiosulfonate bond, and leverage this reactivity using sulfinate-linked probes to enrich and annotate hundreds of endogenous S-nitrosated proteins. In physiological buffers, sulfinic acids do not react with iodoacetamide or disulfides, enabling selective alkylation of free thiols and site-specific analysis of S-nitrosation. In parallel, S-nitrosothiol-linked probes enable enrichment and detection of endogenous S-sulfinated proteins, confirming that a single sulfinic acid can react with a nitrosothiol to form a thiosulfonate linkage. Using this approach, we find that hydrogen peroxide addition increases S-sulfination of human DJ-1 (PARK7) at Cys106, whereas Cys46 and Cys53 are fully oxidized to sulfonic acids. Comparative gel-based analysis of different mouse tissues reveals distinct profiles for both S-nitrosation and S-sulfination. Quantitative proteomic analysis demonstrates that both S-nitrosation and S-sulfination are widespread, yet exhibit enhanced occupancy on select proteins, including thioredoxin, peroxiredoxins, and other validated redox active proteins. Overall, we present a direct, bidirectional method to profile select redox cysteine modifications based on the unique nucleophilicity of sulfinic acids.
Co-reporter:Jeannie L. Hernandez, Dahvid Davda, Jaimeen D. Majmudar, Sang Joon Won, Ashesh Prakash, Alexandria I. Choi and Brent R. Martin
Molecular BioSystems 2016 vol. 12(Issue 6) pp:1799-1808
Publication Date(Web):18 Mar 2016
DOI:10.1039/C6MB00019C
Epithelial cells form spatially-organized adhesion complexes that establish polarity gradients, regulate cell proliferation, and direct wound healing. As cells accumulate oncogenic mutations, these key tumor suppression mechanisms are disrupted, eliminating many adhesion complexes and bypassing contact inhibition. The transcription factor Snail is often expressed in malignant cancers, where it promotes transcriptional reprogramming to drive epithelial–mesenchymal transition (EMT) and establishes a more invasive state. S-Palmitoylation describes the fatty-acyl post-translational modification of cysteine residues in proteins, and is required for membrane anchoring, trafficking, localization and function of hundreds of proteins involved in cell growth, polarity, and signaling. Since Snail-expression disrupts apico-basolateral cell polarity, we asked if Snail-dependent transformation induces proteome-wide changes in S-palmitoylation. MCF10A breast cancer cells were retrovirally transduced with Snail and correlated proteome-wide changes in protein abundance and S-palmitoylation were profiled by using stable isotope labeling in cell culture with amino acid (SILAC) mass spectrometry. This analysis identified increased levels of proteins involved in migration, glycolysis, and cell junction remodeling, and decreased levels of proteins involved in cell adhesion. Overall, protein S-palmitoylation is highly correlated with protein abundance, yet for a subset of proteins, this correlation is uncoupled. These findings suggest that Snail-overexpression affects the S-palmitoylation cycle of some proteins, which may participate in cell polarity and tumor suppression.
Co-reporter:Hao Xu; Jaimeen D. Majmudar; Dahvid Davda; Phani Ghanakota; Ki H. Kim; Heather A. Carlson; Hollis D. Showalter; Brent R. Martin;Gordon L. Amidon
Molecular Pharmaceutics 2015 Volume 12(Issue 9) pp:3399-3407
Publication Date(Web):August 11, 2015
DOI:10.1021/acs.molpharmaceut.5b00414
Understanding the mechanistic basis of prodrug delivery and activation is critical for establishing species-specific prodrug sensitivities necessary for evaluating preclinical animal models and potential drug–drug interactions. Despite significant adoption of prodrug methodologies for enhanced pharmacokinetics, functional annotation of prodrug activating enzymes is laborious and often unaddressed. Activity-based protein profiling (ABPP) describes an emerging chemoproteomic approach to assay active site occupancy within a mechanistically similar enzyme class in native proteomes. The serine hydrolase enzyme family is broadly reactive with reporter-linked fluorophosphonates, which have shown to provide a mechanism-based covalent labeling strategy to assay the activation state and active site occupancy of cellular serine amidases, esterases, and thioesterases. Here we describe a modified ABPP approach using direct substrate competition to identify activating enzymes for an ethyl ester prodrug, the influenza neuraminidase inhibitor oseltamivir. Substrate-competitive ABPP analysis identified carboxylesterase 1 (CES1) as an oseltamivir-activating enzyme in intestinal cell homogenates. Saturating concentrations of oseltamivir lead to a four-fold reduction in the observed rate constant for CES1 inactivation by fluorophosphonates. WWL50, a reported carbamate inhibitor of mouse CES1, blocked oseltamivir hydrolysis activity in human cell homogenates, confirming CES1 is the primary prodrug activating enzyme for oseltamivir in human liver and intestinal cell lines. The related carbamate inhibitor WWL79 inhibited mouse but not human CES1, providing a series of probes for analyzing prodrug activation mechanisms in different preclinical models. Overall, we present a substrate-competitive activity-based profiling approach for broadly surveying candidate prodrug hydrolyzing enzymes and outline the kinetic parameters for activating enzyme discovery, ester prodrug design, and preclinical development of ester prodrugs.
Co-reporter:Dahvid Davda and Brent R. Martin
MedChemComm 2014 vol. 5(Issue 3) pp:268-276
Publication Date(Web):02 Dec 2013
DOI:10.1039/C3MD00333G
Protein palmitoylation describes the hydrophobic post-translational modification of cysteine residues in certain proteins, and is required for the spatial organization and composition of cellular membrane environments. Certain palmitoylated proteins are processed by acyl protein thioesterase (APT) enzymes, which catalyze thioester hydrolysis of palmitoylated cysteine residues. Inhibiting APT enzymes disrupts Ras trafficking and attenuates oncogenic growth signaling, highlighting these enzymes as potential therapeutic targets. As members of the serine hydrolase enzyme family, APT enzymes can be assayed by fluorophosphonate activity-based protein profiling (ABPP) methods, allowing rapid profiling of inhibitor selectivity and potency. In this review, we discuss recent progress in the development of potent and selective inhibitors to APT enzymes, including both reversible and irreversible inhibitor chemotypes. These examples highlight how ABPP methods integrate with medicinal chemistry for the discovery and optimization of inhibitors in complex proteomes.
Co-reporter:Brent R. Martin
Biopolymers 2014 Volume 101( Issue 2) pp:131-132
Publication Date(Web):
DOI:10.1002/bip.22370
No abstract is available for this article.
Co-reporter:Jaimeen D. Majmudar ;Brent R. Martin
Biopolymers 2014 Volume 101( Issue 2) pp:173-179
Publication Date(Web):
DOI:10.1002/bip.22342
ABSTRACT
Cysteine is a uniquely reactive amino acid, capable of undergoing both nucleophlilic and oxidative post-translational modifications. One such oxidation reaction involves the covalent modification of cysteine via the gaseous second messenger nitric oxide (NO), termed S-nitrosylation (SNO). This dynamic post-translational modification is involved in the redox regulation of proteins across all phylogenic kingdoms. In mammals, calcium-dependent activation of NO synthase triggers the local release of NO, which activates nearby guanylyl cyclases and cGMP-dependent pathways. In parallel, diffusible NO can locally modify redox active cellular thiols, functionally modulating many redox sensitive enzymes. Aberrant SNO is implicated in the pathology of many diseases, including neurodegeneration, inflammation, and stroke. In this review, we discuss current methods to label sites of SNO for biochemical analysis. The most popular method involves a series of biochemical steps to mask free thiols followed by selective nitrosothiol reduction and capture. Other emerging methods include mechanism-based phosphine probes and mercury enrichment chemistry. By bridging new enrichment approaches with high-resolution mass spectrometry, large-scale analysis of protein nitrosylation has highlighted new pathways of oxidative regulation. © 2013 Wiley Periodicals, Inc. Biopolymers 101: 173–179, 2014.
Co-reporter:Dahvid Davda, Mahmoud A. El Azzouny, Christopher T.M.B. Tom, Jeannie L. Hernandez, Jaimeen D. Majmudar, Robert T. Kennedy, and Brent R. Martin
ACS Chemical Biology 2013 Volume 8(Issue 9) pp:1912
Publication Date(Web):July 11, 2013
DOI:10.1021/cb400380s
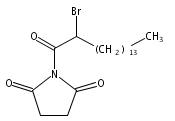
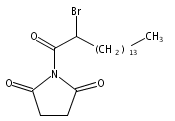
2-Bromohexadecanoic acid, or 2-bromopalmitate, was introduced nearly 50 years ago as a nonselective inhibitor of lipid metabolism. More recently, 2-bromopalmitate re-emerged as a general inhibitor of protein S-palmitoylation. Here, we investigate the cellular targets of 2-bromopalmitate through the synthesis and application of click-enabled analogues. In cells, 2-bromopalmitate is converted to 2-bromopalmitoyl-CoA, although less efficiently than free palmitate. Once conjugated to CoA, probe reactivity is dramatically enhanced. Importantly, both 2-bromopalmitate and 2-bromopalmitoyl-CoA label DHHC palmitoyl acyl transferases (PATs), the enzymes that catalyze protein S-palmitoylation. Mass spectrometry analysis of enriched 2-bromopalmitate targets identified PAT enzymes, transporters, and many palmitoylated proteins, with no observed preference for CoA-dependent enzymes. These data question whether 2-bromopalmitate (or 2-bromopalmitoyl-CoA) blocks S-palmitoylation by inhibiting protein acyl transferases, or by blocking palmitate incorporation by direct covalent competition. Overall, these findings highlight the promiscuous reactivity of 2BP and validate clickable 2BP analogues as activity-based probes of diverse membrane associated enzymes.
Co-reporter:Christopher T.M.B. Tom and Brent R. Martin
ACS Chemical Biology 2013 Volume 8(Issue 1) pp:46
Publication Date(Web):December 7, 2012
DOI:10.1021/cb300607e
Protein palmitoylation describes the post-translational fatty acyl thioesterification of cellular cysteine residues and is critical for the localization, trafficking, and compartmentalization of a large number of membrane proteins. This labile thioester modification facilitates a dynamic acylation cycle that directionally traffics key signaling complexes, receptors, and channels to select membrane compartments. Chemical enrichment and advanced mass spectrometry-based proteomics methods have highlighted a pervasive role for palmitoylation across all eukaryotes, including animals, plants, and parasites. Emerging chemical tools promise to open new avenues to study the enzymes, substrates, and dynamics of this distinct post-translational modification.
Co-reporter:Christopher T. M. B. Tom, John E. Crellin, Hashim F. Motiwala, Matthew B. Stone, Dahvid Davda, William Walker, Yu-Hsuan Kuo, Jeannie L. Hernandez, Kristin J. Labby, Lyanne Gomez-Rodriguez, Paul M. Jenkins, Sarah L. Veatch and Brent R. Martin
Chemical Communications 2017 - vol. 53(Issue 53) pp:NaN7388-7388
Publication Date(Web):2017/06/07
DOI:10.1039/C7CC02285A
Here we report a ratiometric fluorescent probe for chemoselective conjugation to sulfenic acids in living cells. Our approach couples an α-fluoro-substituted dimedone to an aminonaphthalene fluorophore (F-DiNap), which upon sulfenic acid conjugation is locked as the 1,3-diketone, changing the fluorophore excitation. F-DiNap reacts with S-sulfenylated proteins at equivalent rates to current probes, but the α-fluorine substitution blocks side-reactions with biological aldehydes.
.jpg)






























![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7.png)
![Ethanamine, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/114300/134179-38-7_b.png)