Co-reporter:Yaozong Chen;Yiping Jiang;Zhihong Guo
Biochemistry December 6, 2016 Volume 55(Issue 48) pp:6685-6695
Publication Date(Web):November 9, 2016
DOI:10.1021/acs.biochem.6b00889
o-Succinylbenzoyl-CoA (OSB-CoA) synthetase, or MenE, catalyzes an essential step in vitamin K biosynthesis and is a valuable drug target. Like many other adenylating enzymes, it changes its structure to accommodate substrate binding, catalysis, and product release along the path of a domain alternation catalytic mechanism. We have determined the crystal structure of its complex with the adenylation product, o-succinylbenzoyl-adenosine monophosphate (OSB-AMP), and captured a new postadenylation state. This structure presents unique features such as a strained conformation for the bound adenylate intermediate to indicate that it represents the enzyme state after completion of the adenylation reaction but before release of the C domain in its transition to the thioesterification conformation. By comparison to the ATP-bound preadenylation conformation, structural changes are identified in both the reactants and the active site to allow inference about how these changes accommodate and facilitate the adenylation reaction and to directly support an in-line backside attack nucleophilic substitution mechanism for the first half-reaction. Mutational analysis suggests that the conserved His196 plays an important role in desolvation of the active site rather than stabilizing the transition state of the adenylation reaction. In addition, comparison of the new structure with a previously determined OSB-AMP-bound structure of the same enzyme allows us to propose a release mechanism of the C domain in its alteration to form the thioesterification conformation. These findings allow us to better understand the domain alternation catalytic mechanism of MenE as well as many other adenylating enzymes.
Co-reporter:Haigang Song; Chen Dong; Mingming Qin; Yaozong Chen; Yueru Sun; Jingjing Liu; Wan Chan;Zhihong Guo
Journal of the American Chemical Society 2016 Volume 138(Issue 23) pp:7244-7247
Publication Date(Web):May 23, 2016
DOI:10.1021/jacs.6b03437
Enamine is a well-known reactive intermediate mediating essential thiamine-dependent catalysis in central metabolic pathways. However, this intermediate is not found in the thiamine-dependent catalysis of the vitamin K biosynthetic enzyme MenD. Instead, an active tetrahedral post-decarboxylation intermediate is stably formed in the enzyme and was structurally determined at 1.34 Å resolution in crystal. This intermediate takes a unique conformation that allows only one proton between its tetrahedral reaction center and the exo-ring nitrogen atom of the aminopyrimidine moiety in the cofactor with a short distance of 3.0 Å. It is readily convertible to the final product of the enzymic reaction with a solvent-exchangeable proton at its reaction center. These results show that the thiamine-dependent enzyme utilizes a tetrahedral intermediate in a mechanism distinct from the enamine catalytic chemistry.
Co-reporter:Jie Shi, Xinyun Cao, Yaozong Chen, John E. Cronan, and Zhihong Guo
Biochemistry 2016 Volume 55(Issue 48) pp:
Publication Date(Web):November 7, 2016
DOI:10.1021/acs.biochem.6b00818
Pimeloyl-acyl carrier protein (ACP) methyl esterase is an α/β-hydrolase that catalyzes the last biosynthetic step of pimeloyl-ACP, a key intermediate in biotin biosynthesis. Intriguingly, multiple nonhomologous isofunctional forms of this enzyme that lack significant sequence identity are present in diverse bacteria. One such esterase, Escherichia coli BioH, has been shown to be a typical α/β-hydrolase fold enzyme. To gain further insights into the role of this step in biotin biosynthesis, we have determined the crystal structure of another widely distributed pimeloyl-ACP methyl esterase, Haemophilus influenzae BioG, at 1.26 Å. The BioG structure is similar to the BioH structure and is composed of an α-helical lid domain and a core domain that contains a central seven-stranded β-pleated sheet. However, four of the six α-helices that flank both sides of the BioH core β-sheet are replaced with long loops in BioG, thus forming an unusual α/β-hydrolase fold. This structural variation results in a significantly decreased thermal stability of the enzyme. Nevertheless, the lid domain and the residues at the lid–core interface are well conserved between BioH and BioG, in which an analogous hydrophobic pocket for pimelate binding as well as similar ionic interactions with the ACP moiety are retained. Biochemical characterization of site-directed mutants of the residues hypothesized to interact with the ACP moiety supports a similar substrate interaction mode for the two enzymes. Consequently, these enzymes package the identical catalytic function under a considerably different protein surface.
Co-reporter:Haigang Song;Ri Xu ; Zhihong Guo
ChemBioChem 2015 Volume 16( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/cbic.201490068
Co-reporter:Haigang Song;Ri Xu ; Zhihong Guo
ChemBioChem 2015 Volume 16( Issue 1) pp:100-109
Publication Date(Web):
DOI:10.1002/cbic.201402489
Abstract
CalE6 is a previously uncharacterized protein involved in the biosynthesis of calicheamicins in Micromonospora echinospora. It is a pyridoxal-5′-phosphate-dependent enzyme and exhibits high sequence homology to cystathionine γ-lyases and cystathionine γ-synthases. However, it was found to be active towards methionine and to convert this amino acid into α-ketobutyrate, ammonium, and methanethiol. The crystal structure of the cofactor-bound holoenzyme was resolved at 2.0 Å; it contains two active site residues, Gly105 and Val322, specific for methionine γ-lyases. Modeling of methionine into the active site allows identification of the active site residues responsible for substrate recognition and catalysis. These findings support that CalE6 is a putative methionine γ-lyase producing methanethiol as a building block in biosynthesis of calicheamicins.
Co-reporter:MinJiao Chen;Ming Jiang;ZhiHong Guo
Science China Chemistry 2013 Volume 56( Issue 3) pp:312-320
Publication Date(Web):2013 March
DOI:10.1007/s11426-012-4792-6
The thiamine-dependent enzyme (1R, 2S, 5S, 6S)-2-succinyl-5-enolpyruvyl-6-hydroxyl-3-cyclohexene-1-carboxylate (SEPHCHC) synthase, also known as MenD, catalyzes a Stetter-like reaction in the biosynthesis of vitamin K. It is found to catalyze a novel reductive C-N bond ligation reaction between nitroarenes and α-ketoacids to form N-hydroxamates. This reaction likely proceeds through an enzyme-mediated, slow two-electron reduction of the nitroalkanes to form a nitroso intermediate, which serves as the electrophilic acceptor of the ketoacid-derived acyl anion. The involvement of the nitroso intermediate is supported by the fact that similar N-hydroxamates are readily formed at a much higher rate when nitroso compounds replace the nitro substrates in the chemoenzymatic reactions. These results demonstrate that the thiamine-dependent enzyme is able to catalyze novel, nonnative reactions that may find new chemoenzymatic applications.
Co-reporter:Yueru Sun, Haigang Song, Jie Li, Ming Jiang, Yan Li, Jiahai Zhou, and Zhihong Guo
Biochemistry 2012 Volume 51(Issue 22) pp:4580-4589
Publication Date(Web):May 18, 2012
DOI:10.1021/bi300486j
1,4-Dihydroxy-2-naphthoyl coenzyme A (DHNA-CoA) synthase, or MenB, catalyzes a carbon–carbon bond formation reaction in the biosynthesis of both vitamin K1 and K2. Bicarbonate is crucial to the activity of a large subset of its orthologues but lacks a clearly defined structural and mechanistic role. Here we determine the crystal structure of the holoenzymes from Escherichia coli at 2.30 Å and Synechocystis sp. PCC6803 at 2.04 Å, in which the bicarbonate cofactor is bound to the enzyme active site at a position equivalent to that of the side chain carboxylate of an aspartate residue conserved among bicarbonate-insensitive DHNA-CoA synthases. Binding of the planar anion involves both nonspecific electrostatic attraction and specific hydrogen bonding and hydrophobic interactions. In the absence of bicarbonate, the anion binding site is occupied by a chloride ion or nitrate, an inhibitor directly competing with bicarbonate. These results provide a solid structural basis for the bicarbonate dependence of the enzymatic activity of type I DHNA-CoA synthases. The unique location of the bicarbonate ion in relation to the expected position of the substrate α-proton in the enzyme’s active site suggests a critical catalytic role for the anionic cofactor as a catalytic base in enolate formation.
Co-reporter:HaiGang Song;ZhiHong Guo
Science China Chemistry 2012 Volume 55( Issue 1) pp:98-105
Publication Date(Web):2012 January
DOI:10.1007/s11426-011-4448-y
The gene product Sll1127 is a predicted 1,4-dihydroxy-2-naphthoyl-CoA synthase catalyzing an intramolecular Claisen condensation in the phylloquinone biosynthesis of the cyanobacterium Synechocystis sp. PCC 6803. This predicted catalytic function has been verified and the enzyme has been characterized for the first time with kcat= 0.013 s−1 and KM = 9 μM. Its catalytic activity is found to strictly depend on externally added bicarbonate with an apparent KD = 0.60 mM. In addition, this enzyme is inhibited by its 1,4-dihydroxy-2-naphthoyl-CoA product through high-affinity binding, which causes a 18 nm shift of the inhibitor absorption at 392 to 410 nm and engenders a new absorption peak at 345 nm. All these properties of the cyanobacterial enzyme are closely similar to those of the Escherichia coli orthologue from the menaquinone biosynthetic pathway. These results provide additional supporting evidence for the essential role of bicarbonate as a catalytic base in the enzymatic reaction and the eubacterial origin of the enzymes in the cyanobacterial biosynthesis of phylloquinone.
Co-reporter:Minjiao Chen, Ming Jiang, Yueru Sun, Zu-Feng Guo, and Zhihong Guo
Biochemistry 2011 Volume 50(Issue 26) pp:
Publication Date(Web):May 31, 2011
DOI:10.1021/bi200376x
1,4-Dihydroxy-2-naphthoyl-coenzyme A (DHNA-CoA) synthase, or MenB, catalyzes an intramolecular Claisen condensation involving two oxyanion intermediates in the biosynthetic pathway of menaquinone, an essential respiration electron transporter in many microorganisms. Here we report the finding that the DHNA-CoA product and its analogues bind and inhibit the synthase from Escherichia coli with significant ultraviolet–visible spectral changes, which are similar to the changes induced by deprotonation of the free inhibitors in a basic solution. Dissection of the structure–affinity relationships of the inhibitors identifies the hydroxyl groups at positions 1 (C1-OH) and 4 (C4-OH) of DHNA-CoA or their equivalents as the dominant and minor sites, respectively, for the enzyme–ligand interaction that polarizes or deprotonates the bound ligands to cause the observed spectral changes. In the meantime, spectroscopic studies with active site mutants indicate that C4-OH of the enzyme-bound DHNA-CoA interacts with conserved polar residues Arg-91, Tyr-97, and Tyr-258 likely through a hydrogen bonding network that also includes Ser-161. In addition, site-directed mutation of the conserved Asp-163 to alanine causes a complete loss of the ligand binding ability of the protein, suggesting that the Asp-163 side chain is most likely hydrogen-bonded to C1-OH of DHNA-CoA to provide the dominant polarizing effect. Moreover, this mutation also completely eliminates the enzyme activity, strongly supporting the possibility that the Asp-163 side chain provides a strong stabilizing hydrogen bond to the tetrahedral oxyanion, which takes a position similar to that of C1-OH of the enzyme-bound DHNA-CoA and is the second high-energy intermediate in the intracellular Claisen condensation reaction. Interestingly, both Arg-91 and Tyr-97 are located in a disordered loop forming part of the active site of all available DHNA-CoA synthase structures. Their involvement in the interaction with the small molecule ligands suggests that the disordered loop is folded in interaction with the substrates or reaction intermediates, supporting an induced-fit catalytic mechanism for the enzyme.
Co-reporter:Dr. Richard K. Haynes;Kwan-Wing Cheu;Maggie Mei-Ki Tang;Min-Jiao Chen;Dr. Zu-Feng Guo;Dr. Zhi-Hong Guo;Dr. Paolo Coghi;Dr. Diego Monti
ChemMedChem 2011 Volume 6( Issue 2) pp:279-291
Publication Date(Web):
DOI:10.1002/cmdc.201000508
Abstract
Flavin adenine dinucleotide (FAD) is reduced by NADPH–E. coli flavin reductase (Fre) to FADH2 in aqueous buffer at pH 7.4 under argon. Under the same conditions, FADH2 in turn cleanly reduces the antimalarial drug methylene blue (MB) to leucomethylene blue. The latter is rapidly re-oxidized by artemisinins, thus supporting the proposal that MB exerts its antimalarial activity, and synergizes the antimalarial action of artemisinins, by interfering with redox cycling involving NADPH reduction of flavin cofactors in parasite flavin disulfide reductases. Direct treatment of the FADH2 generated from NADPH–Fre–FAD by artemisinins and antimalaria-active tetraoxane and trioxolane structural analogues under physiological conditions at pH 7.4 results in rapid reduction of the artemisinins, and efficient conversion of the peroxide structural analogues into ketone products. Comparison of the relative rates of FADH2 oxidation indicate optimal activity for the trioxolane. Therefore, the rate of intraparastic redox perturbation will be greatest for the trioxolane, and this may be significant in relation to its enhanced in vitro antimalarial activities. 1H NMR spectroscopic studies using the BNAH–riboflavin (RF) model system indicate that the tetraoxane is capable of using both peroxide units in oxidizing the RFH2 generated in situ. Use of the NADPH–Fre–FAD catalytic system in the presence of artemisinin or tetraoxane confirms that the latter, in contrast to artemisinin, consumes two reducing equivalents of NADPH. None of the processes described herein requires the presence of ferrous iron. Ferric iron, given its propensity to oxidize reduced flavin cofactors, may play a role in enhancing oxidative stress within the malaria parasite, without requiring interaction with artemisinins or peroxide analogues. The NADPH–Fre–FAD system serves as a convenient mimic of flavin disulfide reductases that maintain redox homeostasis in the malaria parasite.
Co-reporter:Zu-Feng Guo, Ming Jiang, Suilan Zheng, Zhihong Guo
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 13) pp:3855-3858
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmcl.2010.05.051
Significant conformational change is detected by circular dichroism and fluorimetry for the major component of the enterobactin synthetase in crowded solutions mimicking the intracellular environment. The structural change correlates well with the extent of the crowding-induced side product suppression in nonribosomal enterobactin synthesis. In contrast, protein-stabilizing solvophobic agents such as glycerol have no effect on the formation of side products, excluding crowding-induced protein stability as a cause for the observed enhancement of the product specificity of the synthetase. These results strongly support that macromolecular crowding is an indispensable physiological factor for normal functioning of the nonribosomal enterobactin synthetase by altering the active sites to increase its product specificity.Macromolecular crowding is found to change the structure of the nonribosomal enterobactin synthetase to increase its product specificity.
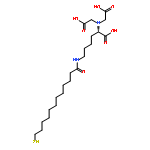
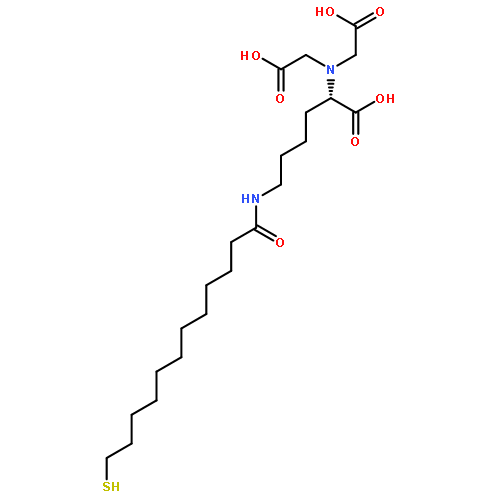
Co-reporter:Xiaolei Chen;Dr. Zu-Feng Guo;Pok Man Lai; Kong Hung Sze; Zhihong Guo
Angewandte Chemie International Edition 2010 Volume 49( Issue 43) pp:7926-7928
Publication Date(Web):
DOI:10.1002/anie.201003369
Co-reporter:Zu-Feng Guo, Yueru Sun, Suilan Zheng and Zhihong Guo
Biochemistry 2009 Volume 48(Issue 8) pp:
Publication Date(Web):February 4, 2009
DOI:10.1021/bi802165x
The type II thioesterase EntH is a hotdog fold protein required for optimal nonribosomal biosynthesis of enterobactin in Escherichia coli. Its proposed proofreading activity in the biosynthesis is confirmed by its efficient restoration of enterobactin synthesis blocked in vitro by analogs of the cognate precursor 2,3-dihydroxybenzoate. Steady-state kinetic studies show that EntH recognizes the phosphopantetheine group and the pattern of hydroxylation in the aryl moiety of its thioester substrates. Remarkably, it is able to distinguish aberrant intermediates from the normal one in the enterobactin assembly line by demonstrating at least 10-fold higher catalytic efficiency toward thioesters derived from aberrant aryl precursors without a para-hydroxyl group, such as salicylate. By structural comparison and site-directed mutagenesis, the thioesterase is found to possess an active site closely resembling that of the 4-hydroxybenzoyl-CoA thioesterase from Arthrobacter sp. strain SU and to involve an acidic residue (glutamate-63) as the catalytic base or nucleophile like all other hotdog thioesterases. In addition, the EntH specificities toward the substrate hydroxylation pattern are found to depend on the active-site histidine-54, threonine-64, serine-67, and methionine-68 with the selectivity significantly reduced or even reversed when they are individually replaced by alanine. These residues are likely responsible for differential interaction of the enzyme with the substrates which leads to distinction between the normal and aberrant precursors in the enterobactin assembly line. These results show that the type II thioesterase evolves its distinctive ability to recognize the aberrant intermediates from the versatile catalytic platform of hotdog proteins and suggests an active search mechanism for type II thioesterases in nonribosomal peptide synthesis.
Co-reporter:Ming Jiang, Xiaolei Chen, Xian-Hui Wu, Minjiao Chen, Yun-Dong Wu and Zhihong Guo
Biochemistry 2009 Volume 48(Issue 29) pp:
Publication Date(Web):June 22, 2009
DOI:10.1021/bi900897h
(1R,6R)-2-Succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate (SHCHC) synthase (MenH) is an α/β fold enzyme containing a catalytically essential serine−histidine−aspartate triad typical of serine proteases but catalyzes a pyruvate elimination reaction initiated by α-proton abstraction in the menaquinone biosynthetic pathway of Escherichia coli. In this study, we identify the active site residues in the synthase through sequence analysis and structural modeling and study their mechanistic roles in MenH catalysis. Steady-state kinetic characterization of site-directed mutants of the active site residues shows that three conserved arginine residues (Arg-90, Arg-124, and Arg-168) likely form ionic salt bridges with three carboxylate groups of the substrate in the Michaelis complex and that the side-chain polar groups of the conserved tyrosine (Tyr-85) and tryptophan (Trp-147) residues likely donate hydrogen bonds to form an “oxyanion hole”. In addition, the pH dependence of the MenH kinetic properties reveals a catalytic base with a pKa highly dependent on the hydroxyl group of the triad serine residue in the enzymatic reaction. Moreover, proton inventory experiments demonstrate that the SHCHC synthase adopts one-proton catalysis like many serine proteases. These results allow the proposal of a mechanism in which the histidine residue of the MenH triad serves as a general base catalyst to deprotonate the triad seryl hydroxyl group in the α-proton abstraction from the substrate. As such, the MenH triad performs a simple and fundamental proton transfer reaction occurring repeatedly in the reactions catalyzed by serine proteases and α/β fold hydrolases, suggesting a common evolutionary origin for all serine−histidine−aspartate triads serving different catalytic functions.
Co-reporter:Ming Jiang, Xiaolei Chen, Zu-Feng Guo, Yang Cao, Minjiao Chen and Zhihong Guo
Biochemistry 2008 Volume 47(Issue 11) pp:
Publication Date(Web):February 20, 2008
DOI:10.1021/bi7023755
Menaquinone is a lipid-soluble molecule that plays an essential role as an electron carrier in the respiratory chain of many bacteria. We have previously shown that its biosynthesis in Escherichia coli involves a new intermediate, 2-succinyl-5-enolpyruvyl-6-hydroxy-3-cyclohexene-1-carboxylate (SEPHCHC), and requires an additional enzyme to convert this intermediate into (1R,6R)-2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate (SHCHC). Here, we report the identification and characterization of MenH (or YfbB), an enzyme previously proposed to catalyze a late step in menaquinone biosynthesis, as the SHCHC synthase. The synthase catalyzes a proton abstraction reaction that results in 2,5-elimination of pyruvate from SEPHCHC and the formation of SHCHC. It is an efficient enzyme (kcat/KM = 2.0 × 107 M−1 s−1) that provides a smaller transition-state stabilization than other enzymes catalyzing proton abstraction from carbon acids. Despite its lack of the proposed thioesterase activity, the SHCHC synthase is homologous to the well-characterized C−C bond hydrolase MhpC. The crystallographic structure of the Vibrio cholerae MenH protein closely resembles that of MhpC and contains a Ser-His-Asp triad typical of serine proteases. Interestingly, this triad is conserved in all MenH proteins and is essential for the SHCHC synthase activity. Mutational analysis found that the catalytic efficiency of the E. coli protein is reduced by 1.4 × 103, 2.1 × 105, and 9.3 × 103 folds when alanine replaces serine, histidine, and aspartate of the triad, respectively. These results show that the SHCHC synthase is closely related to α/β hydrolases but catalyzes a reaction mechanistically distinct from all known hydrolase reactions.
Co-reporter:Guiyang Xie, Mahesh Uttamchandani, Grace Y.J Chen, Xianzhang Bu, San San Lin, Ka Man Wong, Weili Yan, Shao Q Yao, Zhihong Guo
Bioorganic & Medicinal Chemistry Letters 2002 Volume 12(Issue 6) pp:989-992
Publication Date(Web):25 March 2002
DOI:10.1016/S0960-894X(02)00067-7
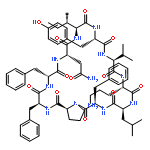
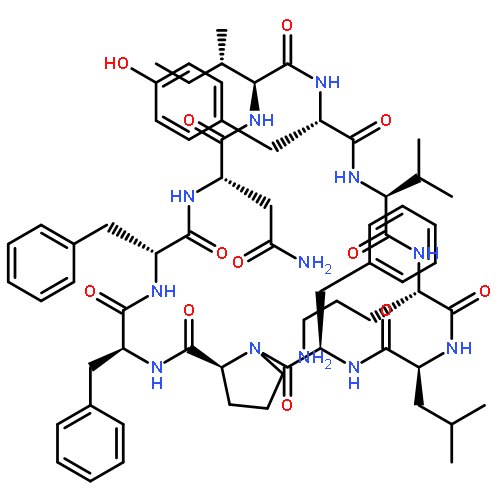
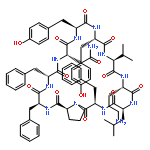
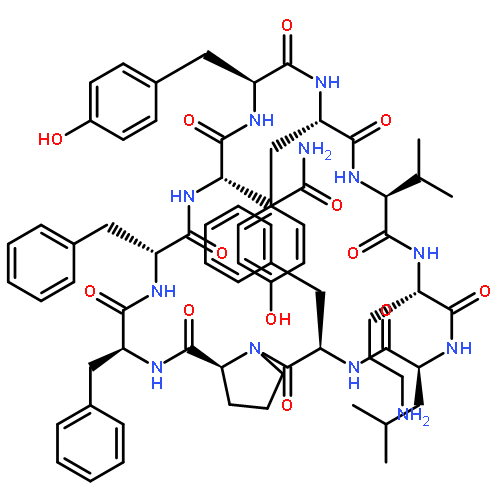
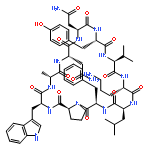
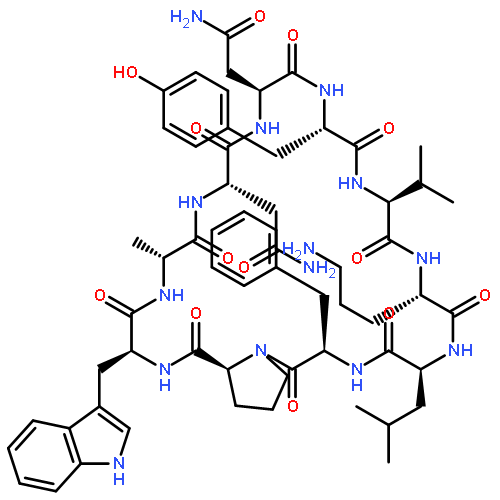
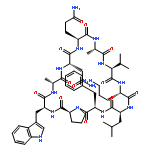
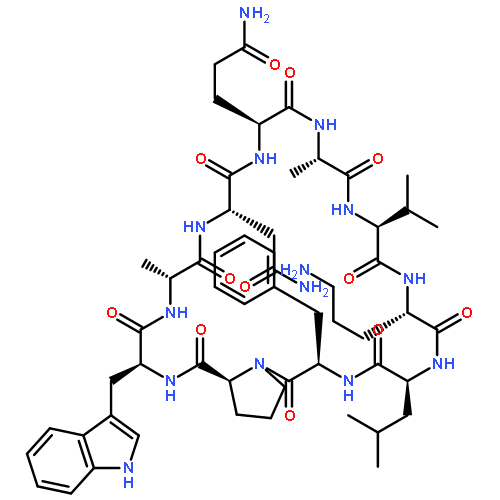
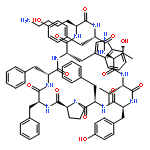
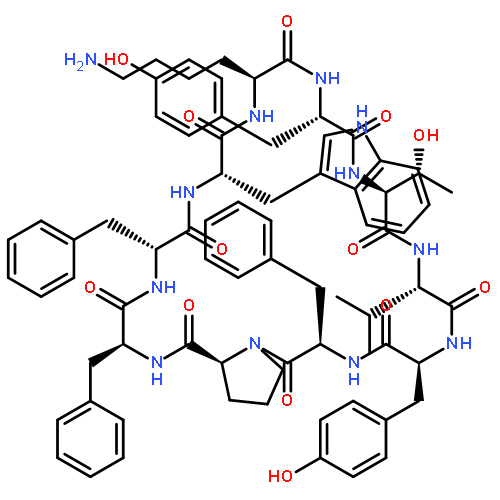
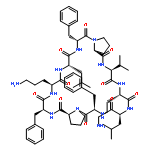
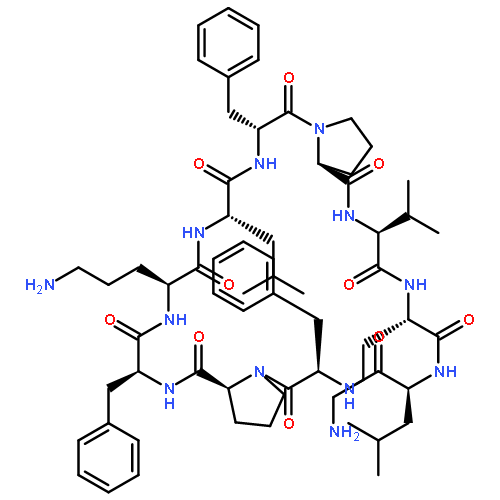
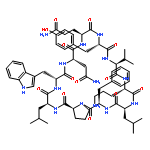
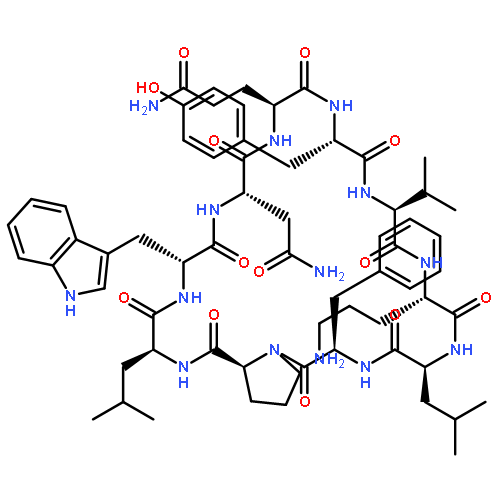
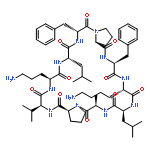
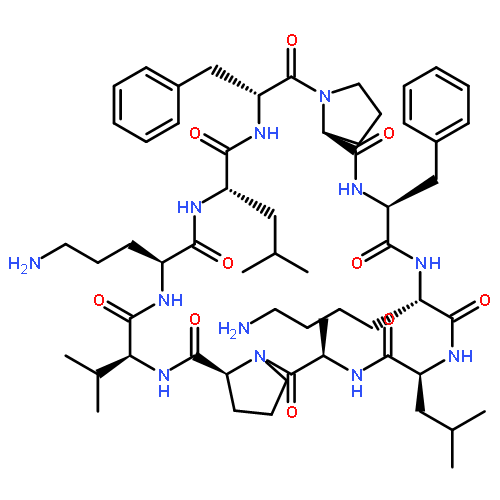
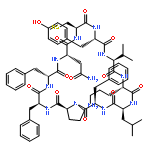
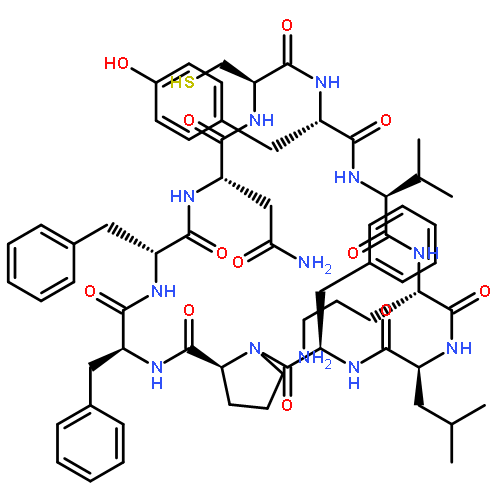
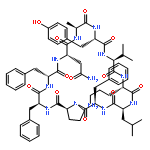
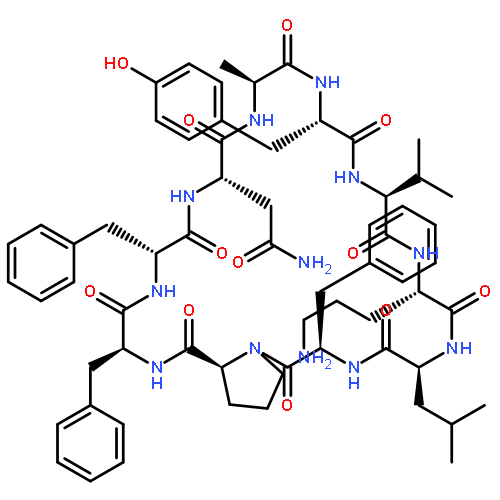
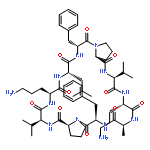
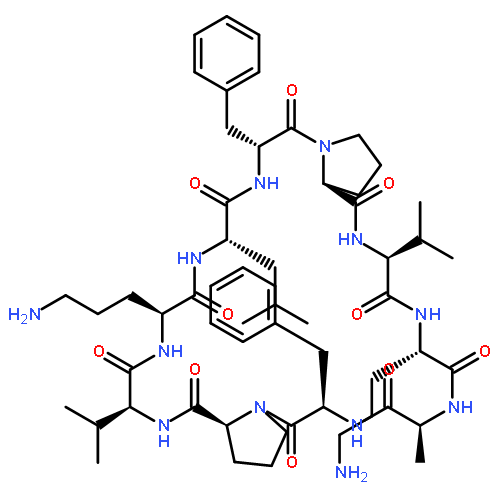
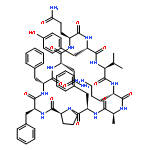
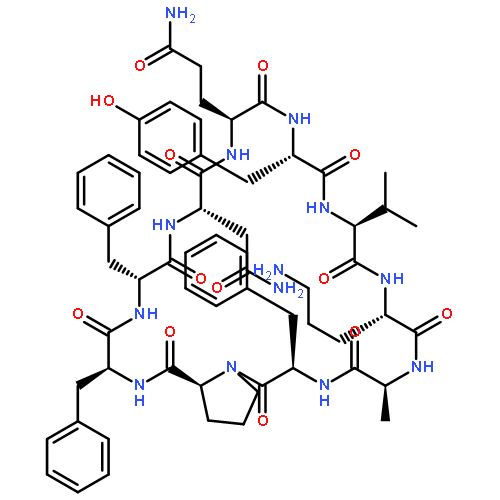
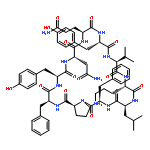
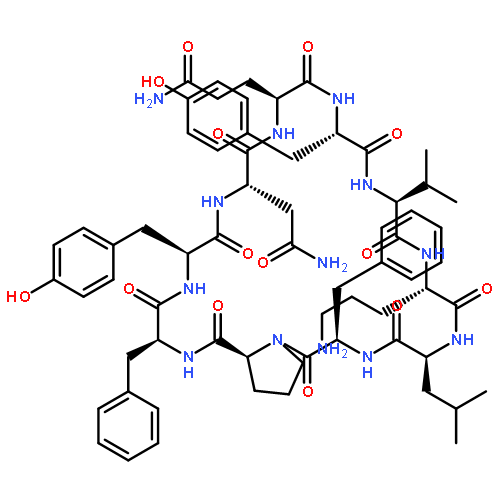
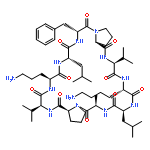
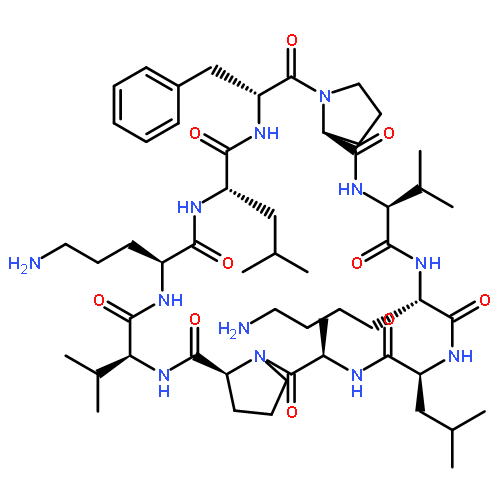
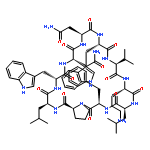
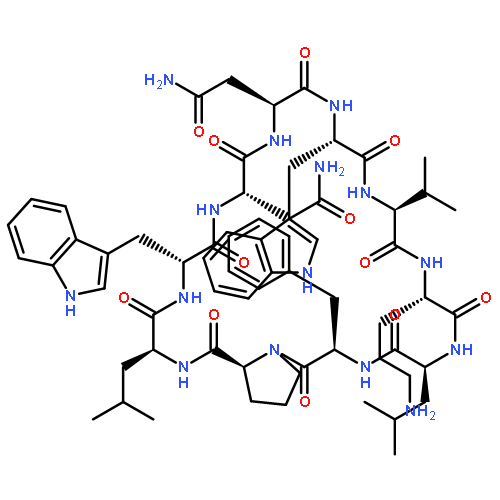
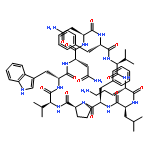
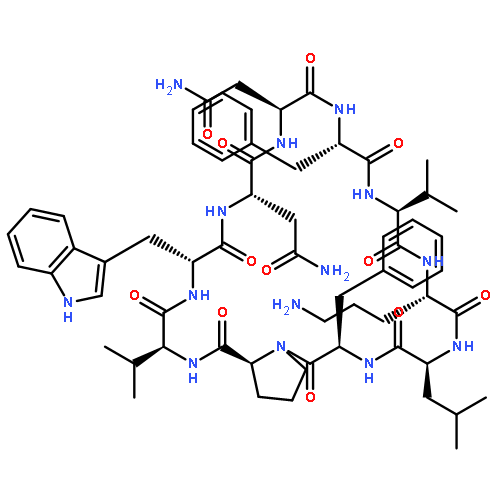
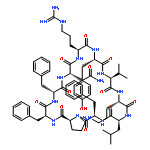
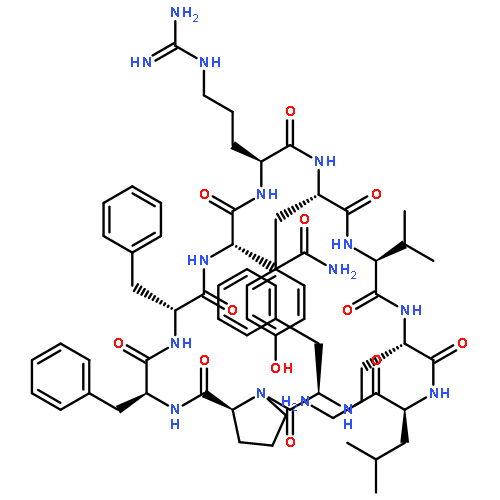
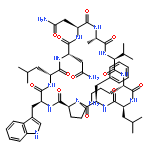
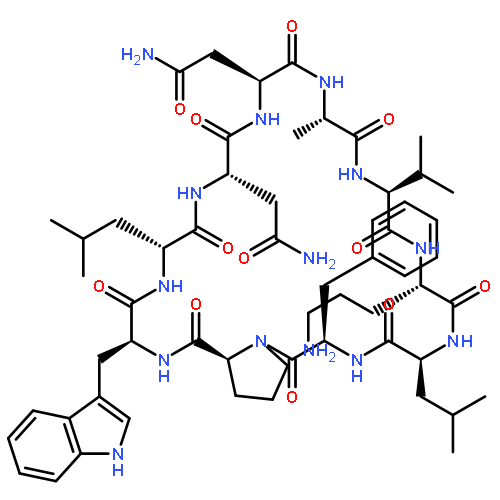
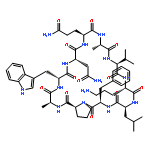
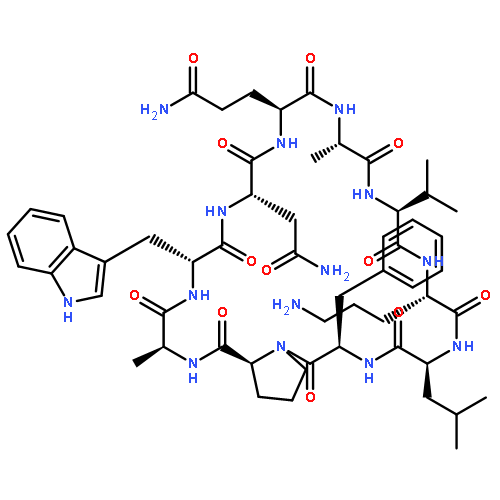
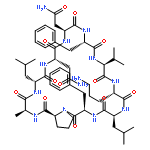
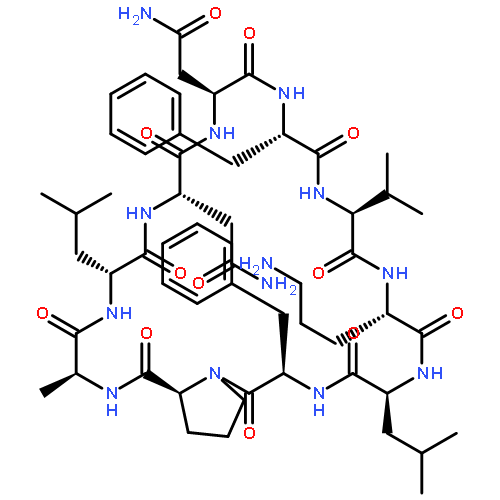
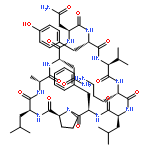
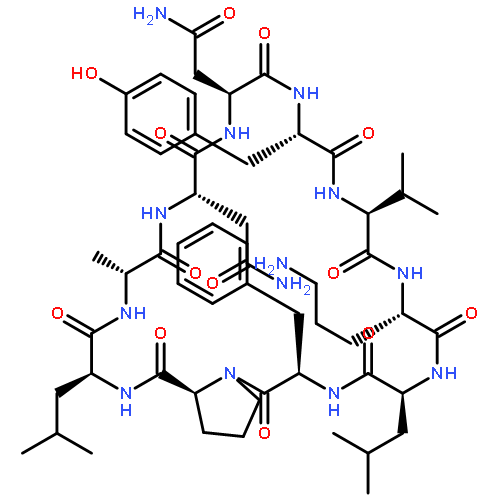
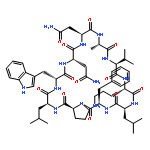
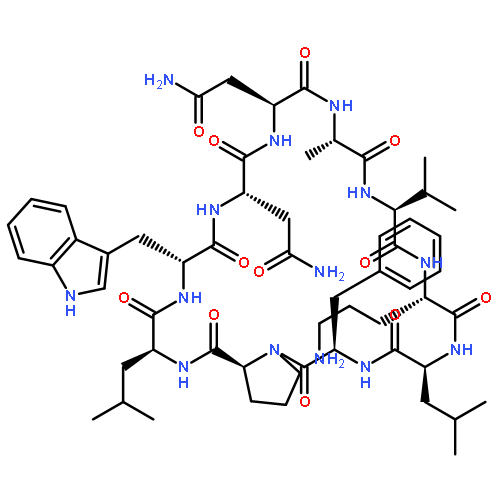
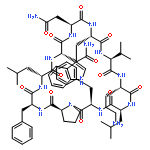
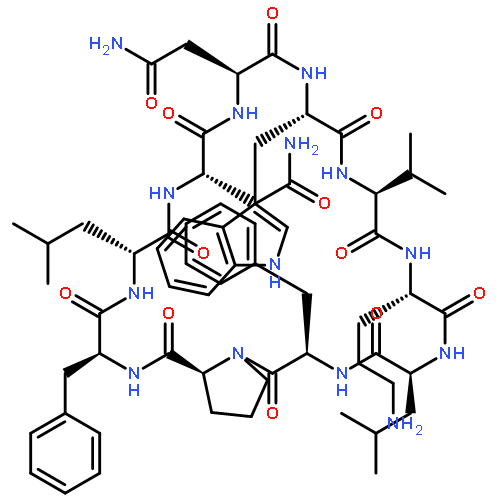
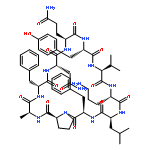
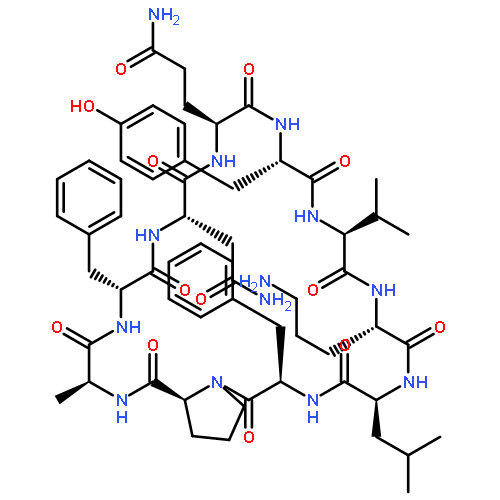
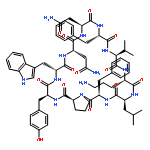
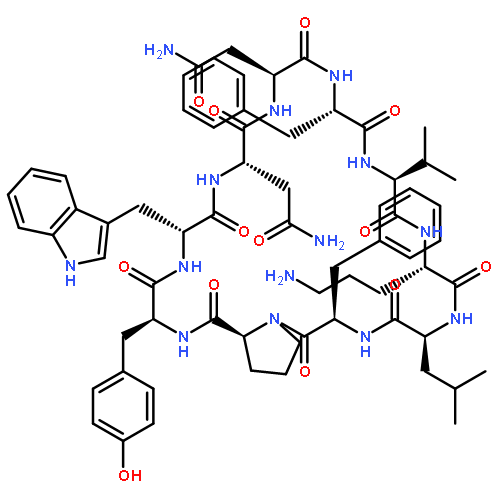
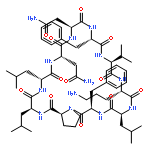
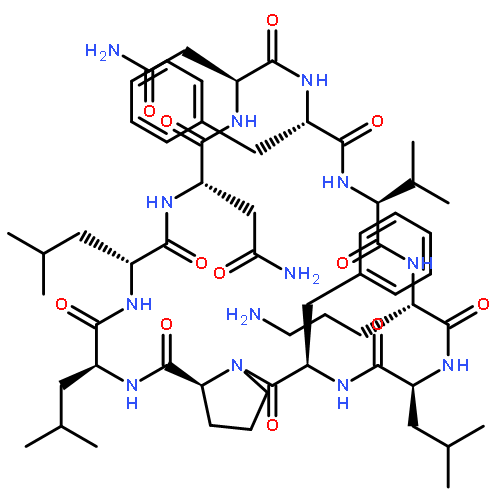
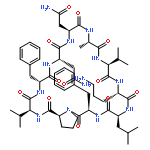
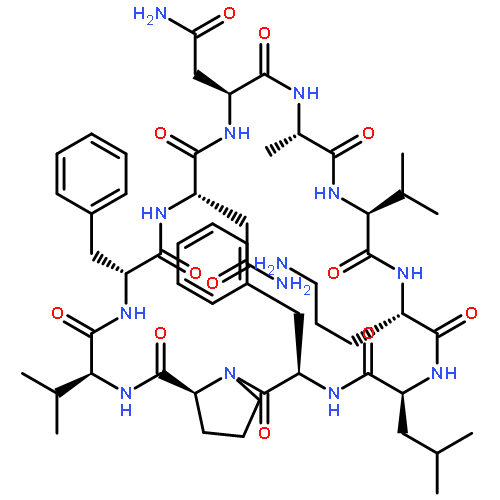
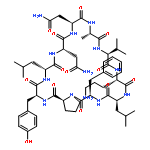
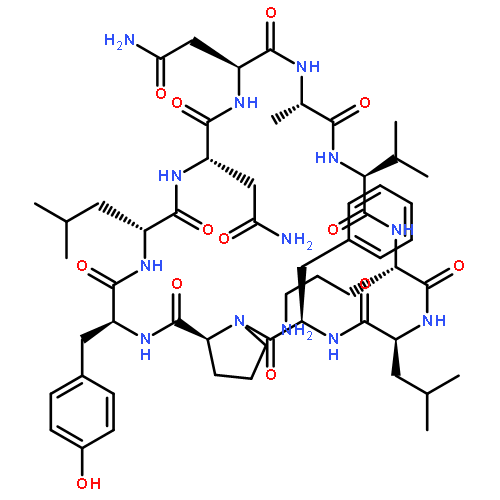
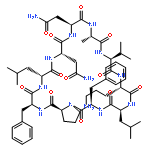
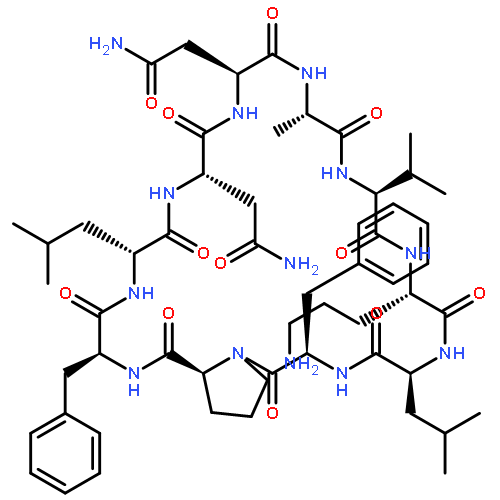
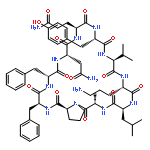
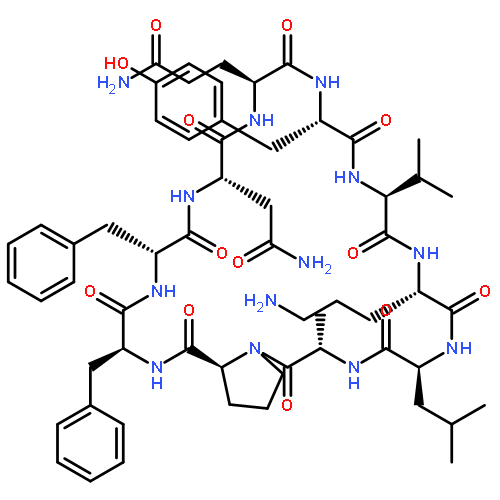
Apparent kinetic constants kcat and Km were determined for tyrocidine thioesterase (TycC TE) using randomized peptide N-acetylcysteamine thioesters as substrate analogues. The enzyme has been found to be adequately active for the synthesis of positional-scanning libraries for novel antibiotic screening with reduced kcat/Km in the range of 2 to 82 folds lower than that of the wild-type sequenceApparent catalyic efficiency of tyrocidine thioesterase towards randomized peptide substrate analogues is reported.

![L-Lysine, N6-[12-(acetylthio)-1-oxododecyl]-N2,N2-bis(carboxymethyl)-](http://img.cochemist.com/ccimg/681300/681239-93-0.png)
![L-Lysine, N6-[12-(acetylthio)-1-oxododecyl]-N2,N2-bis(carboxymethyl)-](http://img.cochemist.com/ccimg/681300/681239-93-0_b.png)


![2,5-Pyrrolidinedione, 1-[(2,4-dihydroxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/186400/186371-01-7.png)
![2,5-Pyrrolidinedione, 1-[(2,4-dihydroxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/186400/186371-01-7_b.png)


![L-LYSINE, N6-[16-(ACETYLTHIO)-1-OXOHEXADECYL]-N2,N2-BIS(CARBOXYMETHYL)-](http://img.cochemist.com/ccimg/681300/681239-92-9.png)
![L-LYSINE, N6-[16-(ACETYLTHIO)-1-OXOHEXADECYL]-N2,N2-BIS(CARBOXYMETHYL)-](http://img.cochemist.com/ccimg/681300/681239-92-9_b.png)

![N-[Nalpha,Nalpha-Bis(carboxymethyl)-L-lysine]-16-mercaptohexadecanamide](http://img.cochemist.com/ccimg/681300/681239-94-1.png)
![N-[Nalpha,Nalpha-Bis(carboxymethyl)-L-lysine]-16-mercaptohexadecanamide](http://img.cochemist.com/ccimg/681300/681239-94-1_b.png)



![2,5-Pyrrolidinedione, 1-[(2,3-dihydroxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/86800/86748-78-9.png)
![2,5-Pyrrolidinedione, 1-[(2,3-dihydroxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/86800/86748-78-9_b.png)
![2,5-Pyrrolidinedione, 1-[(4-hydroxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/70100/70074-31-6.png)
![2,5-Pyrrolidinedione, 1-[(4-hydroxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/70100/70074-31-6_b.png)






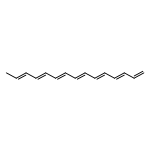
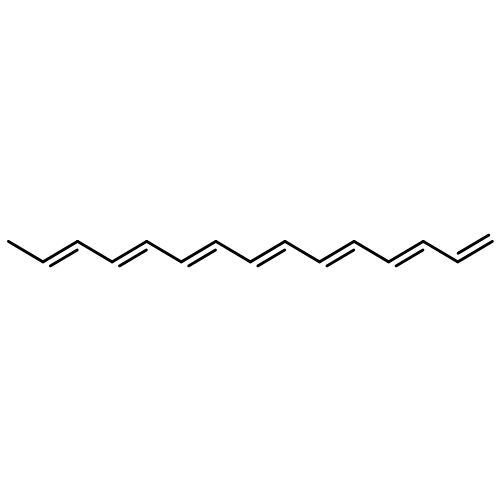
![1,4-Naphthalenedione,2-methyl-3-[(2E,6E,10E,14E)-3,7,11,15,19-pentamethyl-2,6,10,14,18-eicosapentaen-1-yl]-](http://img.cochemist.com/ccimg/1200/1182-68-9.png)
![1,4-Naphthalenedione,2-methyl-3-[(2E,6E,10E,14E)-3,7,11,15,19-pentamethyl-2,6,10,14,18-eicosapentaen-1-yl]-](http://img.cochemist.com/ccimg/1200/1182-68-9_b.png)





![1,3-Cyclohexadiene-1-carboxylicacid, 5-[(1-carboxyethenyl)oxy]-6-hydroxy-, (5S,6S)-](http://img.cochemist.com/ccimg/22700/22642-82-6.png)
![1,3-Cyclohexadiene-1-carboxylicacid, 5-[(1-carboxyethenyl)oxy]-6-hydroxy-, (5S,6S)-](http://img.cochemist.com/ccimg/22700/22642-82-6_b.png)






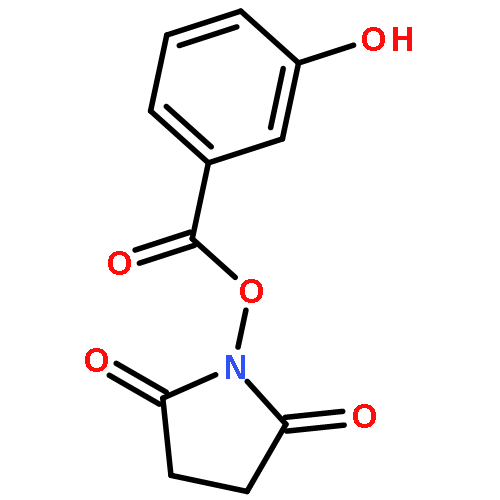
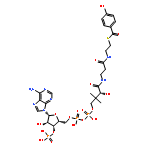

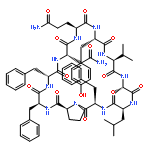
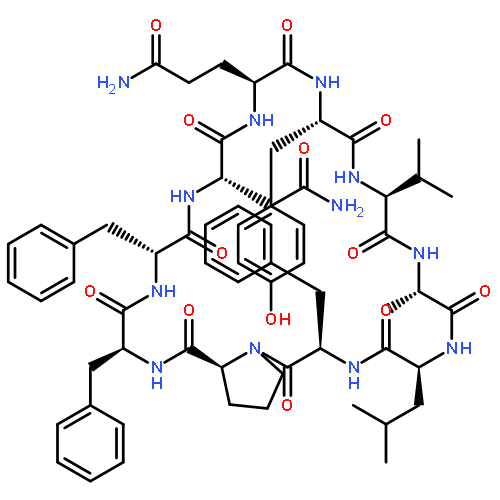
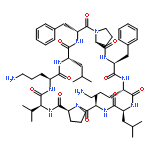
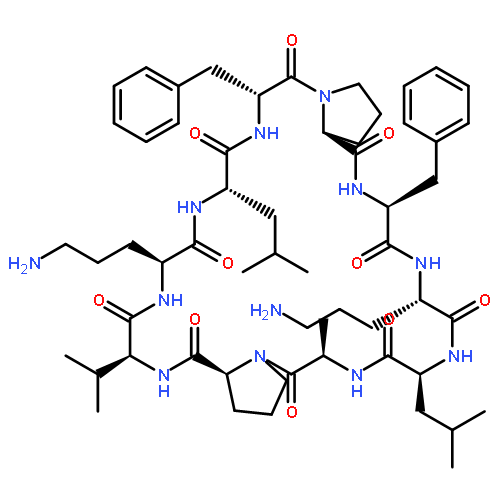
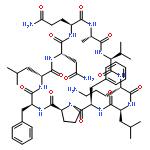
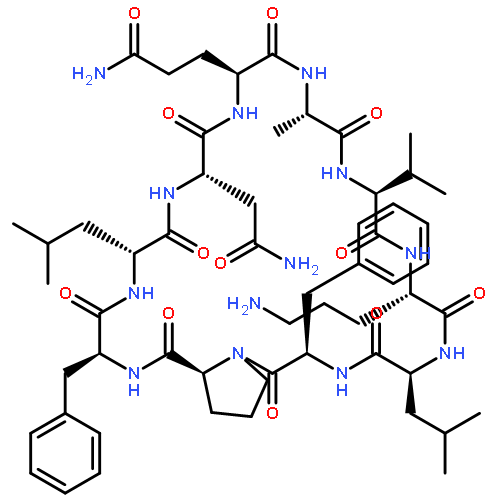
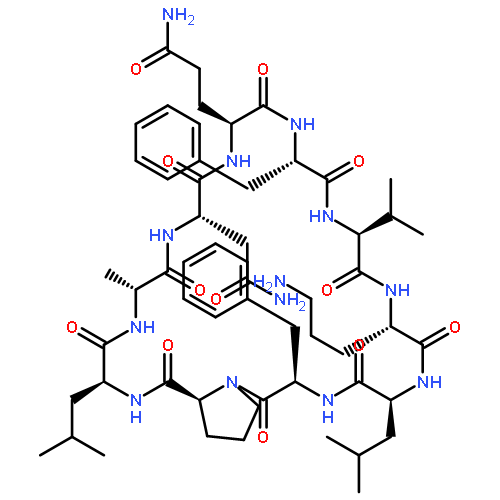
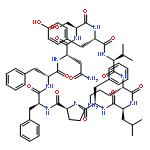
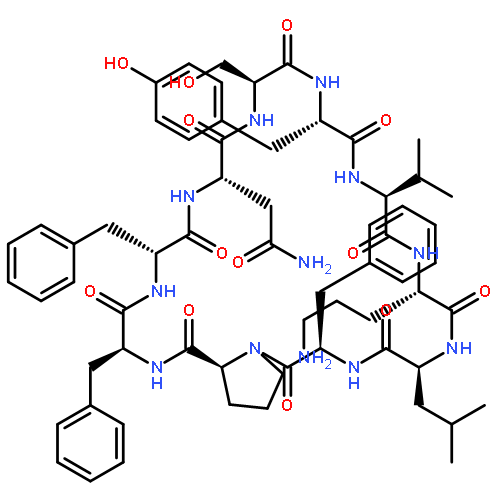
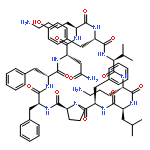
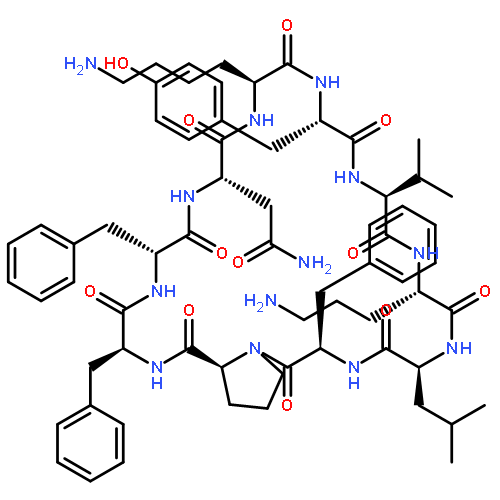
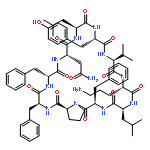
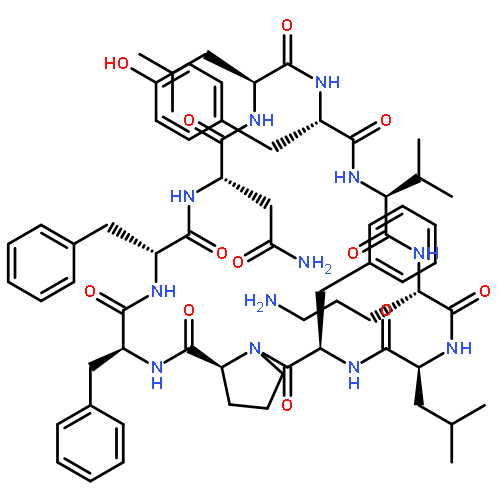
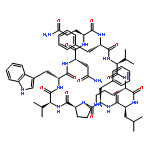
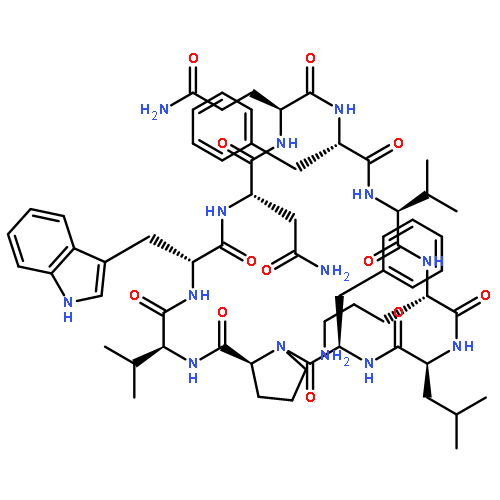
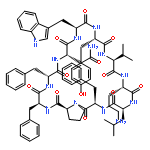
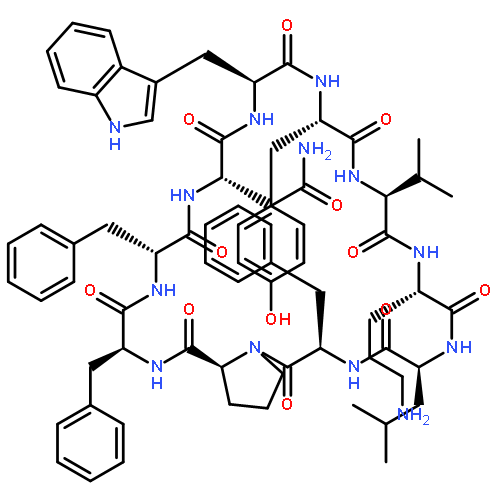
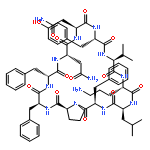
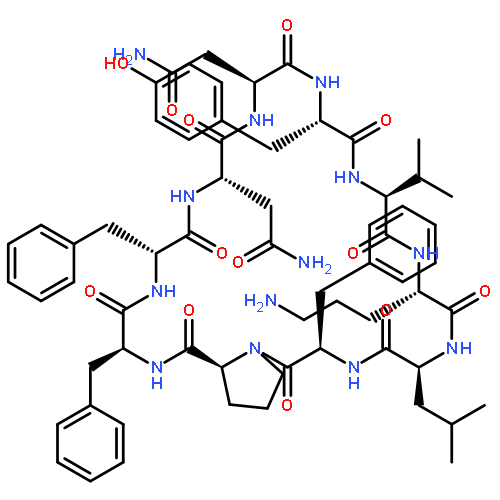
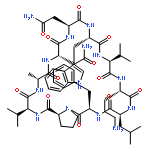
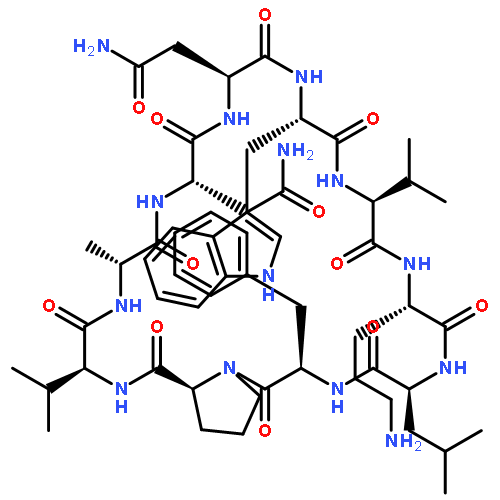
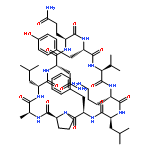
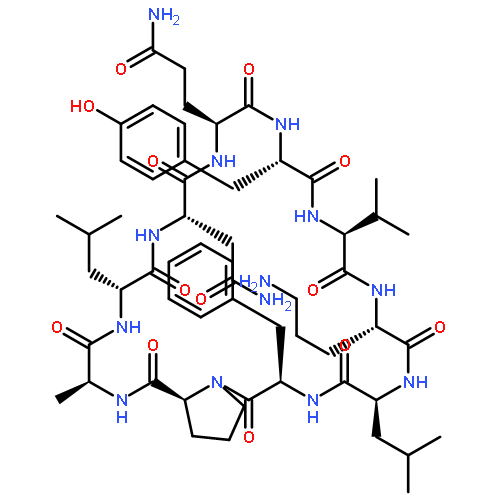
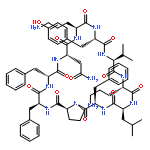
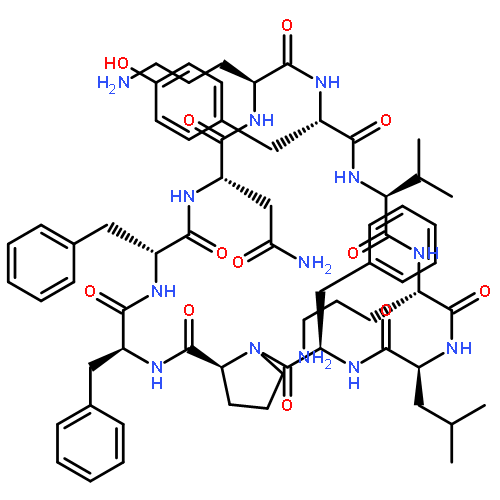
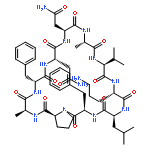
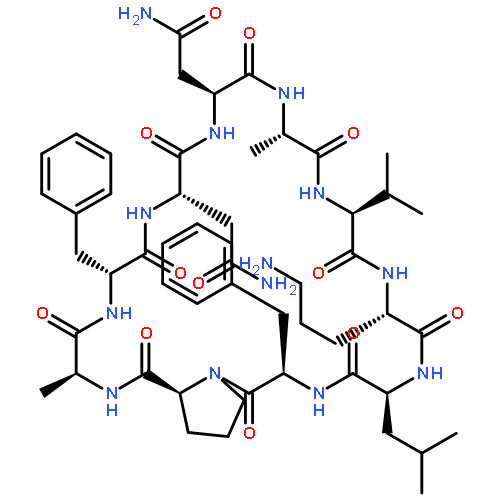
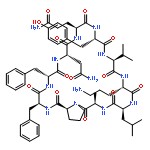
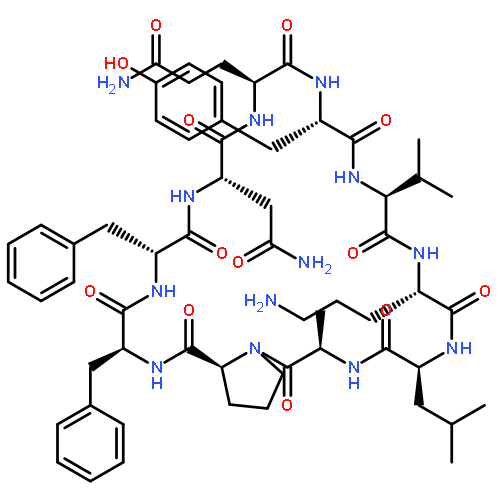
![Carbamic acid,N-[(1R,4Z,8S,13E)-8-[[4,6-dideoxy-4-[[[2,6-dideoxy-4-S-[4-[(6-deoxy-3-O-methyl-a-L-mannopyranosyl)oxy]-3-iodo-5,6-dimethoxy-2-methylbenzoyl]-4-thio-b-D-ribo-hexopyranosyl]oxy]amino]-2-O-[2,4-dideoxy-4-(ethylamino)-3-O-methyl-a-L-threo-pentopyranosyl]-b-D-glucopyranosyl]oxy]-1-hydroxy-13-[2-(methyltrithio)ethylidene]-11-oxobicyclo[7.3.1]trideca-4,9-diene-2,6-diyn-10-yl]-,methyl ester](http://img.cochemist.com/ccimg/108300/108212-75-5.png)
![Carbamic acid,N-[(1R,4Z,8S,13E)-8-[[4,6-dideoxy-4-[[[2,6-dideoxy-4-S-[4-[(6-deoxy-3-O-methyl-a-L-mannopyranosyl)oxy]-3-iodo-5,6-dimethoxy-2-methylbenzoyl]-4-thio-b-D-ribo-hexopyranosyl]oxy]amino]-2-O-[2,4-dideoxy-4-(ethylamino)-3-O-methyl-a-L-threo-pentopyranosyl]-b-D-glucopyranosyl]oxy]-1-hydroxy-13-[2-(methyltrithio)ethylidene]-11-oxobicyclo[7.3.1]trideca-4,9-diene-2,6-diyn-10-yl]-,methyl ester](http://img.cochemist.com/ccimg/108300/108212-75-5_b.png)
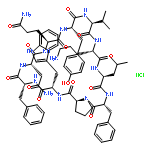
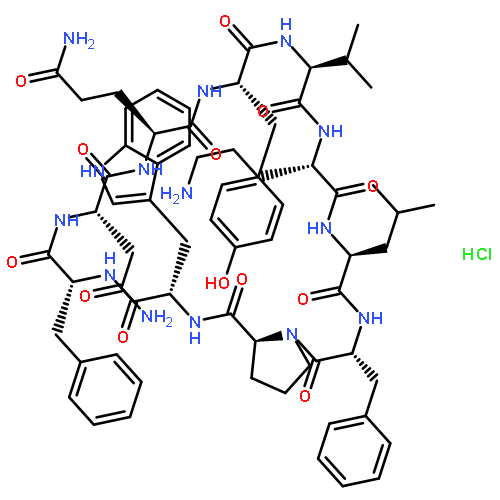




![1,5-Cyclohexadiene-1-carboxylicacid, 3-[(1-carboxyethenyl)oxy]-4-hydroxy-, (3R,4R)-](http://img.cochemist.com/ccimg/700/617-12-9.png)
![1,5-Cyclohexadiene-1-carboxylicacid, 3-[(1-carboxyethenyl)oxy]-4-hydroxy-, (3R,4R)-](http://img.cochemist.com/ccimg/700/617-12-9_b.png)
![Ethanone, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-](http://img.cochemist.com/ccimg/103600/103548-04-5.png)
![Ethanone, 2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-](http://img.cochemist.com/ccimg/103600/103548-04-5_b.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)


![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)
![TERT-BUTYL N-[2-(3,4-DIHYDROXYPHENYL)ETHYL]CARBAMATE](http://img.cochemist.com/ccimg/37100/37034-31-4.png)
![TERT-BUTYL N-[2-(3,4-DIHYDROXYPHENYL)ETHYL]CARBAMATE](http://img.cochemist.com/ccimg/37100/37034-31-4_b.png)
![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5.png)
![Benzamide,N,N',N''-[(3S,7S,11S)-2,6,10-trioxo-1,5,9-trioxacyclododecane-3,7,11-triyl]tris[2,3-dihydroxy-](http://img.cochemist.com/ccimg/28400/28384-96-5_b.png)


















![2-Methyl-2-propanyl N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-tyrosinate](http://img.cochemist.com/ccimg/133900/133852-23-0.png)
![2-Methyl-2-propanyl N-[(9H-fluoren-9-ylmethoxy)carbonyl]-L-tyrosinate](http://img.cochemist.com/ccimg/133900/133852-23-0_b.png)









