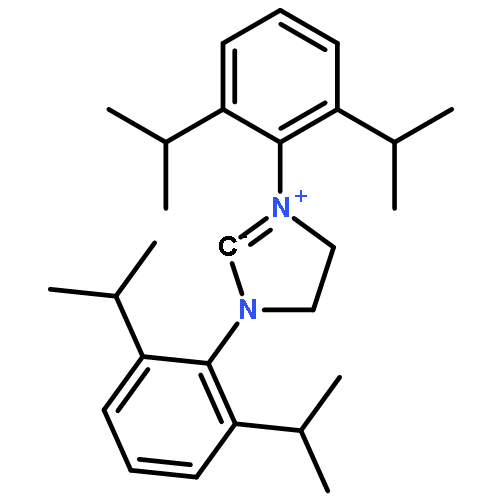
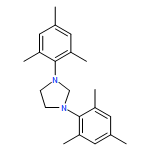
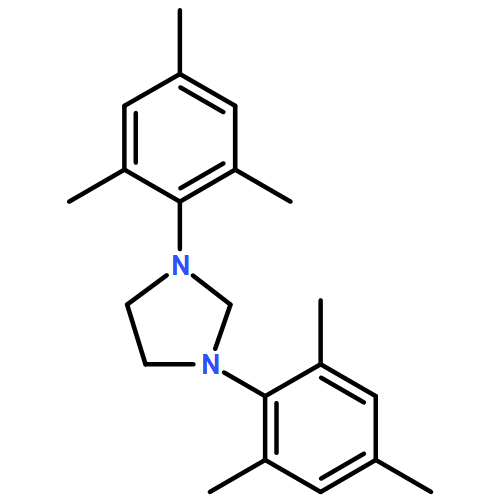
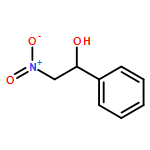
Co-reporter:Daniel von der Heiden, Seyma Bozkus, Martin Klussmann, and Martin Breugst
The Journal of Organic Chemistry April 21, 2017 Volume 82(Issue 8) pp:4037-4037
Publication Date(Web):March 28, 2017
DOI:10.1021/acs.joc.7b00445
Molecular iodine, an easy to handle solid, has been successfully employed as a catalyst in different organic transformations for more than 100 years. Despite being active even in very small amounts, the origin of this remarkable catalytic effect is still unknown. Both a halogen bond mechanism as well as hidden Brønsted acid catalysis are frequently discussed as possible explanations. Our kinetic analyses reveal a reaction order of 1 in iodine, indicating that higher iodine species are not involved in the rate-limiting transition state. Our experimental investigations rule out hidden Brønsted acid catalysis by partial decomposition of I2 to HI and suggest a halogen bond activation instead. Finally, molecular iodine turned out to be a similar if not superior catalyst for Michael additions compared with typical Lewis acids.
Co-reporter:J. Schmauck;M. Breugst
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 38) pp:8037-8045
Publication Date(Web):2017/10/04
DOI:10.1039/C7OB01599B
Pnicogen bonding is a noncovalent interaction between the electrophilic region of a phosphorus atom and a Lewis base. Although this interaction can be comparable in strength to other noncovalent interactions, no systematic application in organic synthesis or catalysis is known so far. To identify the potential of this interaction for organocatalysis, we have now analysed different pnicogen-bond donors as catalysts for the activation of three different model reactions employing density functional theory. Our calculations suggest rate accelerations of several orders of magnitude for all cases indicating that synthetic applications should be feasible. Furthermore, our results indicate that pnicogen-bond donors can be comparable to halogen-bond-based catalysts in these reactions.
Co-reporter:Martin Breugst, Eric Detmar, and Daniel von der Heiden
ACS Catalysis 2016 Volume 6(Issue 5) pp:3203
Publication Date(Web):April 8, 2016
DOI:10.1021/acscatal.6b00447
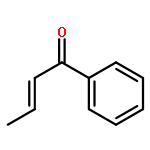
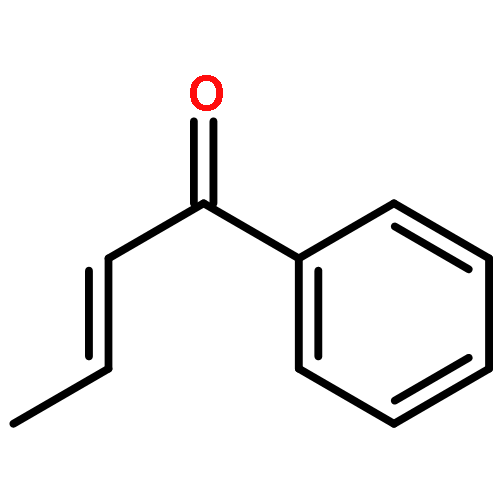
Molecular iodine is an excellent catalyst for many organic transformations, but the origin of its catalytic activity is still unknown. To answer this question, we have analyzed four iodine-catalyzed reactions by density functional theory. Our calculations reveal that molecular iodine significantly reduces the activation free energies (−7.6 < ΔG⧧ < –1.8 kcal mol–1) for reactions involving α,β-unsaturated carbonyls or nitrostyrenes. Closer analysis of the nature of the interaction between iodine and the corresponding Michael acceptors suggests that halogen bonding is the origin of the catalytic activity. The computational and experimental studies show that hidden Brønsted acid catalysis as a competing pathway due to the formation of hydrogen iodide via hypoiodites in aprotic solvents seems less likely for these reactions.Keywords: density functional theory; halogen bonding; iodine catalysis; Michael additions; reaction mechanisms
Co-reporter:Ludovik Noël-Duchesneau, Elodie Lagadic, Fabrice Morlet-Savary, Jean-François Lohier, Isabelle Chataigner, Martin Breugst, Jacques Lalevée, Annie-Claude Gaumont, and Sami Lakhdar
Organic Letters 2016 Volume 18(Issue 22) pp:5900-5903
Publication Date(Web):November 4, 2016
DOI:10.1021/acs.orglett.6b02983
A novel and efficient method for the generation of phosphinoyl radicals from the combination of diphenyliodonium salt (Ph2I+,–OTf) with triethylamine (Et3N) in the presence of secondary phosphine oxides is reported. By employing this practical and simple approach, a large variety of 6-phosphorylated phenanthridines have been synthesized through the addition of phosphinoyl radicals to isonitriles as radical acceptors. The reaction works smoothly in the absence of any transition metal or photocatalyst. On the basis of electron paramagnetic resonance (EPR) and density functional theory (DFT) calculations, the mechanism of this reaction is discussed.
Co-reporter:M.Sc. Marcus Brauns;Frédéric Muller;Daniel Gülden;Dr. Dietrich Böse;Dr. Wolfgang Frey;Dr. Martin Breugst;Dr. Jörg Pietruszka
Angewandte Chemie International Edition 2016 Volume 55( Issue 4) pp:1548-1552
Publication Date(Web):
DOI:10.1002/anie.201509198
Abstract
The use of a convenient protecting group for boronates allows a selective, catalyzed SN2′ reaction to generate allylboronates which are applied for the synthesis of enantiomerically pure homoallylic alcohols. Depending on the configuration of both catalyst and the protecting group any of the four possible stereoisomers can be formed. The rationale behind the selective addition is supported by density functional theory calculations.
Co-reporter:M.Sc. Marcus Brauns;Frédéric Muller;Daniel Gülden;Dr. Dietrich Böse;Dr. Wolfgang Frey;Dr. Martin Breugst;Dr. Jörg Pietruszka
Angewandte Chemie 2016 Volume 128( Issue 4) pp:1574-1578
Publication Date(Web):
DOI:10.1002/ange.201509198
Abstract
Die Verwendung einer einfachen Schutzgruppe für Boronsäureester ermöglicht eine selektive katalysierte SN2′-Reaktion, um Allylboronsäureester zu erzeugen, die zur Synthese von enantiomerenreinen Homoallylalkoholen genutzt werden können. Abhängig von der Konfiguration der Schutzgruppe und der des Katalysators können alle vier möglichen Stereoisomere erhalten werden. Mithilfe von Dichtefunktionalrechnungen wird die Selektivität der Allyladdition erklärt.
Co-reporter:YoungKu Kang, Weijie Chen, Martin Breugst, and Daniel Seidel
The Journal of Organic Chemistry 2015 Volume 80(Issue 19) pp:9628-9640
Publication Date(Web):September 8, 2015
DOI:10.1021/acs.joc.5b01384
Cyclic amines such as 1,2,3,4-tetrahydroisoquinoline undergo regiodivergent annulation reactions with 4-nitrobutyraldehydes. These redox-neutral transformations enable the asymmetric synthesis of highly substituted polycyclic ring systems in just two steps from commercial materials. The utility of this process is illustrated in a rapid synthesis of (−)-protoemetinol. Computational studies provide mechanistic insights and implicate the elimination of acetic acid from an ammonium nitronate intermediate as the rate-determining step.
Co-reporter:Dr. Naeem Gulzar;Dr. Kevin Mark Jones;Hannelore Konnerth;Dr. Martin Breugst;Dr. Martin Klussmann
Chemistry - A European Journal 2015 Volume 21( Issue 8) pp:3367-3376
Publication Date(Web):
DOI:10.1002/chem.201405376
Abstract
The acid-catalyzed reactions of photochemically generated tetrahydrocarbazole peroxides with anilines have been studied experimentally and computationally to identify the underlying reaction mechanism. The kinetic data indicate a reaction order of one in the hydroperoxide and zero in the aniline. Computational investigations using density functional theory support the experimental findings and predict an initial tautomerization between an imine and enamine substructure of the primarily generated tetrahydrocarbazole peroxide to be the rate controlling step. The enamine tautomer then loses hydrogen peroxide upon protonation, generating a stabilized allylic carbocation that is reversibly trapped by solvent or aniline to form the isolated products.
Co-reporter:Martin Breugst and K. N. Houk
The Journal of Organic Chemistry 2014 Volume 79(Issue 13) pp:6302-6309
Publication Date(Web):June 13, 2014
DOI:10.1021/jo501227m
The Henry reaction between benzaldehyde and nitromethane catalyzed by a cyclophane-based bisthiourea has been studied with density functional theory [M06-2X-D3/def2-TZVPP/IEFPCM//TPSS-D2/6-31G(d)/IEFPCM]. The results of our study reveal that the transformation involves the reaction of a thiourea–nitronate complex with the uncoordinated aldehyde. On the basis of our calculations, the formation of the major stereoisomer is kinetically preferred. Employing smaller model systems, we show that the observed stereoselectivity arises primarily from differences in hydrogen bonding in diastereomeric transition states.