Co-reporter:Qing Li, Li YiKam Hon Hoi, Peter Marek, George Georgiou, Brent L. Iverson
ACS Chemical Biology 2017 Volume 12(Issue 2) pp:
Publication Date(Web):December 15, 2016
DOI:10.1021/acschembio.6b00547
An enzyme engineering technology involving yeast endoplasmic reticulum (ER) sequestration screening (YESS) has been recently developed. Here, a new method is established, in which the YESS platform is combined with NextGen sequencing (NGS) to enable a comprehensive survey of protease specificity. In this approach, a combinatorial substrate library is targeted to the yeast ER and transported through the secretory pathway, interacting with any protease(s) residing in the ER. Multicolor FACS screening is used to isolate cells labeled with fluorophore-conjugated antibodies, followed by NGS to profile the cleaved substrates. The YESS-NGS method was successfully applied to profile the sequence specificity of the wild-type and an engineered variant of the tobacco etch mosaic virus protease. Proteolysis in the yeast secretory pathway was also mapped for the first time in vivo revealing a major cleavage pattern of Ali/Leu-X-Lys/Arg-Arg. Here Ali is any small aliphatic residue, but especially Leu. This pattern was verified to be due to the well-known endogenous protease Kex2 after comparison to a newly generated Kex2 knockout strain as well as cleavage of peptides with recombinant Kex2 in vitro. This information is particularly important for those using yeast display technology, as library members with Ali/Leu-X-Lys/Arg-Arg patterns are likely being removed from screens via Kex2 cleavage without the researcher’s knowledge.
Co-reporter:B. A. Ikkanda and B. L. Iverson
Chemical Communications 2016 vol. 52(Issue 50) pp:7752-7759
Publication Date(Web):05 Apr 2016
DOI:10.1039/C6CC01861K
A variety of non-covalent interactions (including hydrogen bonding, ionic interactions, metal coordination and desolvation/solvation) have been utilized to organize oligomers into well-defined structures. Herein is described a survey of aromatic foldamers that capitalize on electrostatic complementarity of substituted aromatic units to drive folding and assembly in aqueous environments. A brief description of recent advances in the understanding of aromatic interactions is provided, followed by examples of foldamers that exploit interactions between aromatic units to drive their assembly in predictable fashion. The history of our aromatic foldamers is traced from the first structure designed to fold into a pleated structure in an aqueous environment to a heteroduplex system more related to nucleic acids. Taken together, the results demonstrate that electrostatic complementarity of aromatic units provides a versatile framework for driving predictable folding and assembly in aqueous environments.
Co-reporter:Cameron Peebles, Christopher D. Wight and Brent L. Iverson
Journal of Materials Chemistry A 2015 vol. 3(Issue 46) pp:12156-12163
Publication Date(Web):01 Oct 2015
DOI:10.1039/C5TC02397A
The stimuli-responsive properties of a series of aromatic conjugated monoalkoxynaphthalene–naphthalimide donor–acceptor dyads were studied. Two of the dyads, dyads 1 and 4, showed a difference in solid-state color between relatively faster (yellow) and slower (yellow-orange or orange) evaporation from solution, while the other dyads, dyad 2 and 3, only showed one color (yellow-green) for both evaporation rates. Importantly, highly solvatochromic dyad 4 displayed thermochromic (orange to yellow), mechanochromic (orange to yellow) and vapochromic (yellow to orange) stimuli-responsive behavior in the solid-state with repeatable cycles of color changing. Structural and spectroscopic studies indicated that the stimuli-responsive behavior of dyad 4 is the result of a 180° molecular rotation wherein the thermodynamically more stable head-to-head stacked orange crystalline solid interconverts with a head-to-tail stacked soft-crystalline yellow mesophase. The thermochromic transition of 4 from a presumably more stable crystalline state (orange) to a metastable soft crystalline mesophase state (yellow) that persists at room temperature unless exposed to solvent vapor is particularly noteworthy.
Co-reporter:Cameron Peebles, Paul M. Alvey, Vincent Lynch, and Brent L. Iverson
Crystal Growth & Design 2014 Volume 14(Issue 1) pp:290-299
Publication Date(Web):November 20, 2013
DOI:10.1021/cg401522v
In order to exploit the use of favorable electrostatic interactions between aromatic units in directing the assembly of donor–acceptor (D–A) dyads, the present work examines the ability of conjugated aromatic D–A dyads with symmetric side chains to exhibit solid-state polymorphism as a function of time during the solid formation process. Four such dyads were synthesized, and their packing in the solid state from either slower (10–20 days) or faster (1–2 days) evaporation from solvent was investigated using single crystal X-ray analysis and powder X-ray diffraction. Two of the dyads exhibited tail-to-tail (A–A) packing upon slower evaporation from solvent and head-to-tail (D–A) packing upon faster evaporation from solvent. A combination of single-crystal analysis and XRD patterns were used to create models, wherein a packing model for the other two dyads is proposed. Our findings suggest that while side chain interactions in asymmetric aromatic dyads can play an important role in enforcing segregated D–A dyad assembly, slowly evaporating symmetrically substituted aromatic dyads allows for favorable electrostatic interactions between the aromatic moieties to facilitate the organization of the dyads in the solid state.
Co-reporter:Brian A. Ikkanda, Stevan A. Samuel, and Brent L. Iverson
The Journal of Organic Chemistry 2014 Volume 79(Issue 5) pp:2029-2037
Publication Date(Web):February 6, 2014
DOI:10.1021/jo402704z
Two novel DNA base surrogate phosphoramidites 1 and 2, based upon relatively electron-rich 1,5-dialkoxynaphthalene (DAN) and relatively electron-deficient 1,4,5,8-naphthalenetetracarboxylic diimide (NDI), respectively, were designed, synthesized, and incorporated into DNA oligonucleotide strands. The DAN and NDI artificial DNA bases were inserted within a three-base-pair region within the interior of a 12-mer oligonucleotide duplex in various sequential arrangements and investigated with CD spectroscopy and UV melting curve analysis. The CD spectra of the modified duplexes indicated B-form DNA topology. Melting curve analyses revealed trends in DNA duplex stability that correlate with the known association of DAN and NDI moieties in aqueous solution as well as the known favorable interactions between NDI and natural DNA base pairs. This demonstrates that DNA duplex stability and specificity can be driven by the electrostatic complementarity between DAN and NDI. In the most favorable case, an NDI–DAN–NDI arrangement in the middle of the DNA duplex was found to be approximately as stabilizing as three A–T base pairs.
Co-reporter:Amy Rhoden Smith
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12783-12789
Publication Date(Web):August 6, 2013
DOI:10.1021/ja4057344
The development of small molecules that bind DNA sequence specifically has the potential to modulate gene expression in a general way. One mode of DNA binding is intercalation, or the insertion of molecules between DNA base pairs. We have developed a modular polyintercalation system in which intercalating naphthalene diimide (NDI) units are connected by flexible linkers that alternate between the minor and major grooves of DNA when bound. We recently reported a threading tetraintercalator with a dissociation half-life of 16 days, the longest reported to date, from its preferred 14 bp binding site. Herein, three new tetraintercalator derivatives were synthesized with one, two, and three additional methylene units in the central major groove-binding linker. These molecules displayed dissociation half-lives of 57, 27, and 18 days, respectively, from the 14 bp site. The optimal major groove-binding linker was used in the design of an NDI hexaintercalator that was analyzed by gel-shift assays, DNase I footprinting, and UV–vis spectroscopy. The hexaintercalator bound its entire 22 bp binding site, the longest reported specific binding site for a synthetic, non-nucleic acid-based DNA binding molecule, but with a significantly faster dissociation rate compared to the tetraintercalators.
Co-reporter:Cameron Peebles;Rebecca Pil ;Dr. Brent L. Iverson
Chemistry - A European Journal 2013 Volume 19( Issue 35) pp:11598-11602
Publication Date(Web):
DOI:10.1002/chem.201302009
Abstract
The thermally induced conformational switching of a stacked dialkxoynaphthalene–naphthalenetetracarboxylic diimide (DAN–NDI) amphiphilic foldamer to an NDI–NDI fibril aggregate is described. The aggregated fibril structures were explored by UV/Vis, circular dichroism (CD), atomic-force microscopy (AFM), and TEM techniques. Our findings indicate that the aromatic DAN–NDI interactions of the original foldamer undergoes transformation to a fibrillar assembly with aromatic NDI–NDI stacked interactions. These structural insights could help inform new molecular designs and increase our understanding of fibrillar assembly and aggregation process in aqueous solution.
Co-reporter:Amy Rhoden Smith, Brian A. Ikkanda, Garen G. Holman, and Brent L. Iverson
Biochemistry 2012 Volume 51(Issue 22) pp:4445-4452
Publication Date(Web):May 3, 2012
DOI:10.1021/bi300317n
Small molecules that bind DNA in a sequence-specific manner could act as antibiotic, antiviral, or anticancer agents because of their potential ability to manipulate gene expression. Our laboratory has developed threading polyintercalators based on 1,4,5,8-naphthalene diimide (NDI) units connected in a head-to-tail fashion by flexible peptide linkers. Previously, a threading tetraintercalator composed of alternating minor–major–minor groove-binding modules was shown to bind specifically to a 14 bp DNA sequence with a dissociation half-life of 16 days [Holman, G. G., et al. (2011) Nat. Chem. 3, 875–881]. Herein are described new NDI-based tetraintercalators with a different major groove-binding module and a reversed N to C directionality of one of the minor groove-binding modules. DNase I footprinting and kinetic analyses revealed that these new tetraintercalators are able to discriminate, by as much as 30-fold, 14 bp DNA binding sites that differ by 1 or 2 bp. Relative affinities were found to correlate strongly with dissociation rates, while overall C2 symmetry in the DNA-binding molecule appeared to contribute to enhanced association rates.
Co-reporter:Cameron Peebles, Christopher D. Wight and Brent L. Iverson
Journal of Materials Chemistry A 2015 - vol. 3(Issue 46) pp:NaN12163-12163
Publication Date(Web):2015/10/01
DOI:10.1039/C5TC02397A
The stimuli-responsive properties of a series of aromatic conjugated monoalkoxynaphthalene–naphthalimide donor–acceptor dyads were studied. Two of the dyads, dyads 1 and 4, showed a difference in solid-state color between relatively faster (yellow) and slower (yellow-orange or orange) evaporation from solution, while the other dyads, dyad 2 and 3, only showed one color (yellow-green) for both evaporation rates. Importantly, highly solvatochromic dyad 4 displayed thermochromic (orange to yellow), mechanochromic (orange to yellow) and vapochromic (yellow to orange) stimuli-responsive behavior in the solid-state with repeatable cycles of color changing. Structural and spectroscopic studies indicated that the stimuli-responsive behavior of dyad 4 is the result of a 180° molecular rotation wherein the thermodynamically more stable head-to-head stacked orange crystalline solid interconverts with a head-to-tail stacked soft-crystalline yellow mesophase. The thermochromic transition of 4 from a presumably more stable crystalline state (orange) to a metastable soft crystalline mesophase state (yellow) that persists at room temperature unless exposed to solvent vapor is particularly noteworthy.
Co-reporter:B. A. Ikkanda and B. L. Iverson
Chemical Communications 2016 - vol. 52(Issue 50) pp:NaN7759-7759
Publication Date(Web):2016/04/05
DOI:10.1039/C6CC01861K
A variety of non-covalent interactions (including hydrogen bonding, ionic interactions, metal coordination and desolvation/solvation) have been utilized to organize oligomers into well-defined structures. Herein is described a survey of aromatic foldamers that capitalize on electrostatic complementarity of substituted aromatic units to drive folding and assembly in aqueous environments. A brief description of recent advances in the understanding of aromatic interactions is provided, followed by examples of foldamers that exploit interactions between aromatic units to drive their assembly in predictable fashion. The history of our aromatic foldamers is traced from the first structure designed to fold into a pleated structure in an aqueous environment to a heteroduplex system more related to nucleic acids. Taken together, the results demonstrate that electrostatic complementarity of aromatic units provides a versatile framework for driving predictable folding and assembly in aqueous environments.
.jpg)






























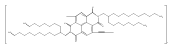
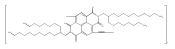
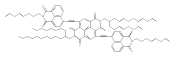
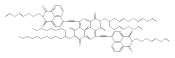














![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-bromo-2-octyl-](/data/chemimg/110700/865443-57-8.png)
![1H-Benz[de]isoquinoline-1,3(2H)-dione, 6-bromo-2-octyl-](/data/chemimg/110700/865443-57-8_b.png)












![2,7-dioctylbenzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone](http://img.cochemist.com/ccimg/34200/34155-21-0.png)
![2,7-dioctylbenzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone](http://img.cochemist.com/ccimg/34200/34155-21-0_b.png)







![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 4,9-dibromo-2,7-bis(2-octyldodecyl)-](/data/chemimg/139000/1100243-35-3.png)
![Benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetrone, 4,9-dibromo-2,7-bis(2-octyldodecyl)-](/data/chemimg/139000/1100243-35-3_b.png)

![2,7-Bis(2-(dimethylamino)ethyl)benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone](/data/chemimg/422600/22291-04-9.png)
![2,7-Bis(2-(dimethylamino)ethyl)benzo[lmn][3,8]phenanthroline-1,3,6,8(2H,7H)-tetraone](/data/chemimg/422600/22291-04-9_b.png)