Co-reporter:Pei Zhou, Jinshi Zhao
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2017 Volume 1862, Issue 11(Issue 11) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.bbalip.2016.11.014
The Raetz pathway of lipid A biosynthesis plays a vital role in the survival and fitness of Gram-negative bacteria. Research efforts in the past three decades have identified individual enzymes of the pathway and have provided a mechanistic understanding of the action and regulation of these enzymes at the molecular level. This article reviews the discovery, biochemical and structural characterization, and regulation of the essential lipid A enzymes, as well as continued efforts to develop novel antibiotics against Gram-negative pathogens by targeting lipid A biosynthesis. This article is part of a Special Issue entitled: Bacterial Lipids edited by Russell E. Bishop
Co-reporter:Qinglin Wu;Benjamin A. Fenton;Jessica L. Wojtaszek
Chemical Communications 2017 vol. 53(Issue 61) pp:8541-8544
Publication Date(Web):2017/07/27
DOI:10.1039/C7CC05021F
CEST-NMR spectroscopy is a powerful tool for probing the conformational dynamics of macromolecules. We present a HNdec-CEST experiment that simplifies the relaxation matrix, reduces fitting parameters, and enhances signal resolution. Importantly, fitting of HNdec-CEST profiles enables robust extraction of exchange rates as well as excited-state chemical shifts and populations.
Co-reporter:Xiaofei Liang, Ramesh Gopalaswamy, Frank Navas III, Eric J. Toone, and Pei Zhou
The Journal of Organic Chemistry 2016 Volume 81(Issue 10) pp:4393-4398
Publication Date(Web):April 29, 2016
DOI:10.1021/acs.joc.6b00589
The difluoromethyl-allo-threonyl hydroxamate-based compound LPC-058 is a potent inhibitor of UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LpxC) in Gram-negative bacteria. A scalable synthesis of this compound is described. The key step in the synthetic sequence is a transition metal/base-catalyzed aldol reaction of methyl isocyanoacetate and difluoroacetone, giving rise to 4-(methoxycarbonyl)-5,5-disubstituted 2-oxazoline. A simple NMR-based determination of enantiomeric purity is also described.
Co-reporter:Chul-Jin Lee, Xiaofei Liang, Ramesh Gopalaswamy, Javaria Najeeb, Eugene D. Ark, Eric J. Toone, and Pei Zhou
ACS Chemical Biology 2014 Volume 9(Issue 1) pp:237
Publication Date(Web):October 11, 2013
DOI:10.1021/cb400067g
The LpxC enzyme in the lipid A biosynthetic pathway is one of the most promising and clinically unexploited antibiotic targets for treatment of multidrug-resistant Gram-negative infections. Progress in medicinal chemistry has led to the discovery of potent LpxC inhibitors with a variety of chemical scaffolds and distinct antibiotic profiles. The vast majority of these compounds, including the nanomolar inhibitors L-161,240 and BB-78485, are highly effective in suppressing the activity of Escherichia coli LpxC (EcLpxC) but not divergent orthologs such as Pseudomonas aeruginosa LpxC (PaLpxC) in vitro. The molecular basis for such promiscuous inhibition of EcLpxC has remained poorly understood. Here, we report the crystal structure of EcLpxC bound to L-161,240, providing the first molecular insight into L-161,240 inhibition. Additionally, structural analysis of the EcLpxC/L-161,240 complex together with the EcLpxC/BB-78485 complex reveals an unexpected backbone flipping of the Insert I βa−βb loop in EcLpxC in comparison with previously reported crystal structures of EcLpxC complexes with l-threonyl-hydroxamate-based broad-spectrum inhibitors. Such a conformational switch, which has only been observed in EcLpxC but not in divergent orthologs such as PaLpxC, results in expansion of the active site of EcLpxC, enabling it to accommodate LpxC inhibitors with a variety of head groups, including compounds containing single (R- or S-enantiomers) or double substitutions at the neighboring Cα atom of the hydroxamate warhead group. These results highlight the importance of understanding inherent conformational plasticity of target proteins in lead optimization.
Co-reporter:Xiaofei Liang ; Chul-Jin Lee ; Jinshi Zhao ; Eric J. Toone
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6954-6966
Publication Date(Web):August 5, 2013
DOI:10.1021/jm4007774
The zinc-dependent deacetylase LpxC catalyzes the committed step of lipid A biosynthesis in Gram-negative bacteria and is a validated target for the development of novel antibiotics to combat multidrug-resistant Gram-negative infections. Many potent LpxC inhibitors contain an essential threonyl-hydroxamate headgroup for high-affinity interaction with LpxC. We report the synthesis, antibiotic activity, and structural and enzymatic characterization of novel LpxC inhibitors containing an additional aryl group in the threonyl-hydroxamate moiety, which expands the inhibitor-binding surface in LpxC. These compounds display enhanced potency against LpxC in enzymatic assays and superior antibiotic activity against Francisella novicida in cell culture. The comparison of the antibiotic activities of these compounds against a leaky Escherichia coli strain and the wild-type strain reveals the contribution of the formidable outer-membrane permeability barrier that reduces the compounds efficacy in cell culture and emphasizes the importance of maintaining a balanced hydrophobicity and hydrophilicity profile in developing effective LpxC-targeting antibiotics.
Co-reporter:Ryan P. Emptage, Charles W. Pemble IV, John D. York, Christian R. H. Raetz, and Pei Zhou
Biochemistry 2013 Volume 52(Issue 13) pp:
Publication Date(Web):March 7, 2013
DOI:10.1021/bi400097z
The sixth step in the lipid A biosynthetic pathway involves phosphorylation of the tetraacyldisaccharide-1-phosphate (DSMP) intermediate by the cytosol-facing inner membrane kinase LpxK, a member of the P-loop-containing nucleoside triphosphate (NTP) hydrolase superfamily. We report the kinetic characterization of LpxK from Aquifex aeolicus and the crystal structures of LpxK in complex with ATP in a precatalytic binding state, the ATP analogue AMP-PCP in the closed catalytically competent conformation, and a chloride anion revealing an inhibitory conformation of the nucleotide-binding P-loop. We demonstrate that LpxK activity in vitro requires the presence of a detergent micelle and formation of a ternary LpxK–ATP/Mg2+–DSMP complex. Using steady-state kinetics, we have identified crucial active site residues, leading to the proposal that the interaction of D99 with H261 acts to increase the pKa of the imidazole moiety, which in turn serves as the catalytic base to deprotonate the 4′-hydroxyl of the DSMP substrate. The fact that an analogous mechanism has not yet been observed for other P-loop kinases highlights LpxK as a distinct member of the P-loop kinase family, a notion that is also reflected through its localization at the membrane, lipid substrate, and overall structure.
Co-reporter:Chul-Jin Lee, Xiaofei Liang, Xin Chen, Daina Zeng, Sang Hoon Joo, Hak Suk Chung, Adam W. Barb, Shauna M. Swanson, Robert A. Nicholas, Yaoxian Li, Eric J. Toone, Christian R.H. Raetz, Pei Zhou
Chemistry & Biology 2011 Volume 18(Issue 1) pp:38-47
Publication Date(Web):28 January 2011
DOI:10.1016/j.chembiol.2010.11.011
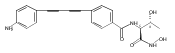
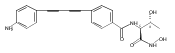
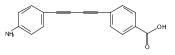
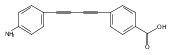
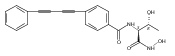
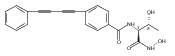
LpxC is an essential enzyme in the lipid A biosynthetic pathway in gram-negative bacteria. Several promising antimicrobial lead compounds targeting LpxC have been reported, though they typically display a large variation in potency against different gram-negative pathogens. We report that inhibitors with a diacetylene scaffold effectively overcome the resistance caused by sequence variation in the LpxC substrate-binding passage. Compound binding is captured in complex with representative LpxC orthologs, and structural analysis reveals large conformational differences that mostly reflect inherent molecular features of distinct LpxC orthologs, whereas ligand-induced structural adaptations occur at a smaller scale. These observations highlight the need for a molecular understanding of inherent structural features and conformational plasticity of LpxC enzymes for optimizing LpxC inhibitors as broad-spectrum antibiotics against gram-negative infections.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (262 K)Download as PowerPoint slideHighlights► LpxC inhibitors with the diacetylene scaffold display a better antibiotic profile ► There exist large inherent conformational differences of individual LpxC orthologs ► Ligand-induced structural adaptation of LpxC occurs at a small scale
Co-reporter:Xiaofei Liang, Chul-Jin Lee, Xin Chen, Hak Suk Chung, Daina Zeng, Christian R.H. Raetz, Yaoxian Li, Pei Zhou, Eric J. Toone
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 2) pp:852-860
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmc.2010.12.017
Compounds inhibiting LpxC in the lipid A biosynthetic pathway are promising leads for novel antibiotics against multidrug-resistant Gram-negative pathogens. We report the syntheses and structural and biochemical characterizations of LpxC inhibitors based on a diphenyl-diacetylene (1,4-diphenyl-1,3-butadiyne) threonyl-hydroxamate scaffold. These studies provide a molecular interpretation for the differential antibiotic activities of compounds with a substituted distal phenyl ring as well as the absolute stereochemical requirement at the C2, but not C3, position of the threonyl group.
Co-reporter:Jie Wen, Jihui Wu, Pei Zhou
Journal of Magnetic Resonance 2011 Volume 209(Issue 1) pp:94-100
Publication Date(Web):March 2011
DOI:10.1016/j.jmr.2010.12.012
Intrinsically disordered proteins (IDPs) play important roles in many critical cellular processes. Due to their limited chemical shift dispersion, IDPs often require four pairs of resonance connectivities (Hα, Cα, Cβ and CO) for establishing sequential backbone assignment. Because most conventional 4-D triple-resonance experiments share an overlapping Cα evolution period, combining existing 4-D experiments does not offer an optimal solution for non-redundant collection of a complete set of backbone resonances. Using alternative chemical shift evolution schemes, we propose a new pair of 4-D triple-resonance experiments – HA(CA)CO(CA)NH/HA(CA)CONH – that complement the 4-D HNCACB/HN(CO)CACB experiments to provide complete backbone resonance information. Collection of high-resolution 4-D spectra with sparse sampling and FFT-CLEAN processing enables efficient acquisition and assignment of complete backbone resonances of IDPs. Importantly, because the CLEAN procedure iteratively identifies resonance signals and removes their associating aliasing artifacts, it greatly reduces the dependence of the reconstruction quality on sampling schemes and produces high-quality spectra even with less-than-optimal sampling schemes.Graphical abstractResearch highlights► Alternative chemical shift evolution schemes are used to construct new experiments. ► 4-D HA(CA)CO(CA)NH/ HA(CA)CONH experiments complement existing experiments. ► Sparse sampling enables efficient data collection for backbone assignment of IDPs. ► FFT-CLEAN produces high-quality spectra even with less-than-optimal sampling schemes.
Co-reporter:Brian E. Coggins, Ronald A. Venters, Pei Zhou
Progress in Nuclear Magnetic Resonance Spectroscopy 2010 Volume 57(Issue 4) pp:381-419
Publication Date(Web):November 2010
DOI:10.1016/j.pnmrs.2010.07.001
Co-reporter:Jon W. Werner-Allen, Brian E. Coggins, Pei Zhou
Journal of Magnetic Resonance 2010 Volume 204(Issue 1) pp:173-178
Publication Date(Web):May 2010
DOI:10.1016/j.jmr.2010.02.017
Amide–amide NOESY provides important distance constraints for calculating global folds of large proteins, especially integral membrane proteins with β-barrel folds. Here, we describe a diagonal-suppressed 4-D NH–NH TROSY-NOESY-TROSY (ds-TNT) experiment for NMR studies of large proteins. The ds-TNT experiment employs a spin state selective transfer scheme that suppresses diagonal signals while providing TROSY optimization in all four dimensions. Active suppression of the strong diagonal peaks greatly reduces the dynamic range of observable signals, making this experiment particularly suitable for use with sparse sampling techniques. To demonstrate the utility of this method, we collected a high resolution 4-D ds-TNT spectrum of a 23 kDa protein using randomized concentric shell sampling (RCSS), and we used FFT-CLEAN processing for further reduction of aliasing artifacts – the first application of these techniques to a NOESY experiment. A comparison of peak parameters in the high resolution 4-D dataset with those from a conventionally-sampled 3-D control spectrum shows an accurate reproduction of NOE crosspeaks in addition to a significant reduction in resonance overlap, which largely eliminates assignment ambiguity. Likewise, a comparison of 4-D peak intensities and volumes before and after application of the CLEAN procedure demonstrates that the reduction of aliasing artifacts by CLEAN does not systematically distort NMR signals.
Co-reporter:Brian E. Coggins, Pei Zhou
Journal of Magnetic Resonance 2007 Volume 184(Issue 2) pp:207-221
Publication Date(Web):February 2007
DOI:10.1016/j.jmr.2006.10.002
We present a novel approach to sampling the NMR time domain, whereby the sampling points are aligned on concentric rings, which we term concentric ring sampling (CRS). Radial sampling constitutes a special case of CRS where each ring has the same number of points and the same relative orientation. We derive theoretically that the most efficient CRS approach is to place progressively more points on rings of larger radius, with the number of points growing linearly with the radius, a method that we call linearly increasing CRS (LCRS). For cases of significant undersampling to reduce measurement time, a randomized LCRS (RLCRS) is also described. A theoretical treatment of these approaches is provided, including an assessment of artifacts and sensitivity. The analytical treatment of sensitivity also addresses the sensitivity of radially sampled data processed by Fourier transform. Optimized CRS approaches are found to produce artifact-free spectra of the same resolution as Cartesian sampling, for the same measurement time. Additionally, optimized approaches consistently yield fewer and smaller artifacts than radial sampling, and have a sensitivity equal to Cartesian and better than radial sampling. We demonstrate the method using numerical simulations, as well as a 3D HNCO experiment on protein G B1 domain.
Co-reporter:Pei Zhou;Christian R. H. Raetz;Ling Jiang;Adam W. Barb
PNAS 2007 Volume 104 (Issue 47 ) pp:18433-18438
Publication Date(Web):2007-11-20
DOI:10.1073/pnas.0709412104
The UDP-3-O-(R-3-hydroxyacyl)-N-acetylglucosamine deacetylase LpxC is an essential enzyme of lipid A biosynthesis in Gram-negative bacteria and a promising
antibiotic target. CHIR-090, the most potent LpxC inhibitor discovered to date, displays two-step time-dependent inhibition
and kills a wide range of Gram-negative pathogens as effectively as ciprofloxacin or tobramycin. In this study, we report
the solution structure of the LpxC–CHIR-090 complex. CHIR-090 exploits conserved features of LpxC that are critical for catalysis,
including the hydrophobic passage and essential active-site residues. CHIR-090 is adjacent to, but does not occupy, the UDP-binding
pocket of LpxC, suggesting that a fragment-based approach may facilitate further optimization of LpxC inhibitors. Additionally,
we identified key residues in the Insert II hydrophobic passage that modulate time-dependent inhibition and CHIR-090 resistance.
CHIR-090 shares a similar, although previously unrecognized, chemical scaffold with other small-molecule antibiotics such
as L-161,240 targeting LpxC, and provides a template for understanding the binding mode of these inhibitors. Consistent with
this model, we provide evidence that L-161,240 also occupies the hydrophobic passage.
Co-reporter:Ming Li;Hemali P. Phatnani;Ziqiang Guan;Harvey Sage;Arno L. Greenleaf;
Proceedings of the National Academy of Sciences 2005 102(49) pp:17636-17641
Publication Date(Web):November 28, 2005
DOI:10.1073/pnas.0506350102
The phosphorylation state of the C-terminal repeat domain (CTD) of the largest subunit of RNA polymerase II changes as polymerase
transcribes a gene, and the distinct forms of the phospho-CTD (PCTD) recruit different nuclear factors to elongating polymerase.
The Set2 histone methyltransferase from yeast was recently shown to bind the PCTD of elongating RNA polymerase II by means
of a novel domain termed the Set2–Rpb1 interacting (SRI) domain. Here, we report the solution structure of the SRI domain
in human Set2 (hSRI domain), which adopts a left-turned three-helix bundle distinctly different from other structurally characterized
PCTD-interacting domains. NMR titration experiments mapped the binding surface of the hSRI domain to helices 1 and 2, and
Biacore binding studies showed that the domain binds preferably to [Ser-2 + Ser-5]-phosphorylated CTD peptides containing
two or more heptad repeats. Point-mutagenesis studies identified five residues critical for PCTD binding. In view of the differential
effects of these point mutations on binding to different CTD phosphopeptides, we propose a model for the hSRI domain interaction
with the PCTD.
Co-reporter:
Nature Structural and Molecular Biology 2003 10(8) pp:645-651
Publication Date(Web):29 June 2003
DOI:10.1038/nsb948
The zinc-dependent UDP-3-O-acyl-N-acetylglucosamine deacetylase (LpxC) catalyzes the first committed step in the biosynthesis of lipid A, the hydrophobic anchor of lipopolysaccharide (LPS) that constitutes the outermost monolayer of Gram-negative bacteria. As LpxC is crucial for the survival of Gram-negative organisms and has no sequence homology to known mammalian deacetylases or amidases, it is an excellent target for the design of new antibiotics. The solution structure of LpxC from Aquifex aeolicus in complex with a substrate-analog inhibitor, TU-514, reveals a novel / fold, a unique zinc-binding motif and a hydrophobic passage that captures the acyl chain of the inhibitor. On the basis of biochemical and structural studies, we propose a catalytic mechanism for LpxC, suggest a model for substrate binding and provide evidence that mobility and dynamics in structural motifs close to the active site have key roles in the capture of the substrate.
Co-reporter:Ming-Tao Pai, Shiou-Ru Tzeng, Jeffrey J. Kovacs, Mignon A. Keaton, ... Pei Zhou
Journal of Molecular Biology (6 July 2007) Volume 370(Issue 2) pp:290-302
Publication Date(Web):6 July 2007
DOI:10.1016/j.jmb.2007.04.015
The BUZ/Znf-UBP domain is a distinct ubiquitin-binding module found in the cytoplasmic deacetylase HDAC6, the E3 ubiquitin ligase BRAP2/IMP, and a subfamily of deubiquitinating enzymes. Here, we report the solution structure of the BUZ domain of Ubp-M, a ubiquitin-specific protease, and its interaction with ubiquitin. Unlike the BUZ domain from isopeptidase T (isoT) that contains a single zinc finger, the Ubp-M BUZ domain features three zinc-binding sites consisting of 12 residues. These zinc ligands form a pair of cross-braced ring fingers encapsulated within a third zinc finger in the primary structure. In contrast to isoT, which can form an N-terminal loop swapped dimer in the crystal state, the formation of additional zinc fingers in the Ubp-M BUZ domain restricts its N-terminal loop to intra-domain interactions. The ubiquitin-binding site of the Ubp-M BUZ domain is mapped to the highly conserved, concave surface formed by the α3 helix and the central β-sheet. We further show that this site binds to the C-terminal tail of free ubiquitin, and corresponding peptides display essentially the same binding affinities as full-length ubiquitin does for the Ubp-M BUZ domain. However, modification of the G76Ub carboxylate group either by a peptide or isopeptide bond abolishes BUZ–domain interaction. The unique ubiquitin-recognition mode of the BUZ domain family suggests that they may function as “sensors” of free ubiquitin in cells to achieve regulatory roles in many aspects of ubiquitin-dependent processes.
Co-reporter:Qinglin Wu, Benjamin A. Fenton, Jessica L. Wojtaszek and Pei Zhou
Chemical Communications 2017 - vol. 53(Issue 61) pp:NaN8544-8544
Publication Date(Web):2017/07/07
DOI:10.1039/C7CC05021F
CEST-NMR spectroscopy is a powerful tool for probing the conformational dynamics of macromolecules. We present a HNdec-CEST experiment that simplifies the relaxation matrix, reduces fitting parameters, and enhances signal resolution. Importantly, fitting of HNdec-CEST profiles enables robust extraction of exchange rates as well as excited-state chemical shifts and populations.