Co-reporter:Meixia Chen, Liang Li, Dafang Zhong, Shuijie Shen, Jiang Zheng, and Xiaoyan Chen
Chemical Research in Toxicology 2016 Volume 29(Issue 2) pp:180
Publication Date(Web):January 13, 2016
DOI:10.1021/acs.chemrestox.5b00427
Retronecine-, otonecine-, and heliotridine-type pyrrolizidine alkaloids (PAs) are all reported to be hepatotoxic. These PAs are suggested to be metabolized to the corresponding electrophilic dehydropyrrolizidine alkaloids (dehydro-PAs) and subsequently conjugated with macromolecules, such as glutathione (GSH). In the present study, a total of five glutathione conjugates, named M1–M5, were detected in rat and human liver microsomal incubations with three retrornecine-type PAs (isoline, retrorsine, or monocrotaline) in the presence of glutathione, and were chemically synthesized. M1 and M3 were unambiguously identified as a pair of epimers of 7-glutathionyl-6,7-dihydro-1-hydroxymethyl-5H-pyrrolizine (7-GSH-DHP), and M4 and M5 were epimers of 7,9-diglutathionyl-6,7-dihydro-1-hydroxymethyl-5H-pyrrolizine (7,9-diGSH-DHP). M2, an extremely unstable conjugate, was proposed to be 9-glutathionyl-6,7-dihydro-1-hydroxymethyl-5H-pyrrolizine (9-GSH-DHP). It was the most abundant among the five GSH conjugates, and the finding corrects the mistake that 7-GSH-DHP is the predominant GSH conjugate derived from dehydro-PAs. Similar patterns in glutathione conjugate profile were observed in the bile of rats treated with the PAs. This is the first study to describe 9-GSH-DHP as a major pyrrolic GSH conjugate of retronecine-type PAs, providing insight into the interactions of dehydro-PAs with biomolecules.
Co-reporter:Yong Yang, Cai Liu, Yifan Zhang, Lei Zhou, Dafang Zhong, Xiaoyan Chen
Journal of Pharmaceutical and Biomedical Analysis 2015 Volume 114() pp:408-415
Publication Date(Web):10 October 2015
DOI:10.1016/j.jpba.2015.05.038
•An ultra-sensitive method for MA bioanalysis with an LLOQ of 10.0 pg/mL.•Pentamethyl derivatives of nitrogen heterocyclic ring-containing BPs was found.•D4-MA exhibited a slower methylation rate than MA to yield pentamethyl derivatives.•The proposed method enabled the ultra-sensitive detection of ZA and RA.•The yield was affected by acidity, exposure time, and anticoagulant.Minodronic acid (MA) is a third-generation bisphosphonate (BP). Its high potency allows lower doses to be administered in clinical settings compared with other BPs, which results in extremely low systemic exposure. Therefore, it is essential to develop an ultra-sensitive bioassay for pharmacokinetics studies of MA. In this work, we used on-cartridge derivatization of MA with trimethylsilyldiazomethane to extract MA from plasma samples and improve its LC–MS/MS behavior. The reaction produced a known derivative, tetramethylated MA, and a novel derivative, pentamethylated MA (PMMA). PMMA exhibited a better signal-to-noise ratio, and was monitored for the quantification of MA. However, the derivatization yield of d4-PMMA was much lower and more variable than that of PMMA, which decreased the effectiveness of its correction function as an internal standard. Therefore, a two-cycle derivatization approach was introduced to increase its yield and improve the reproducibility. The calibration curves of MA showed good linearity over the range of 10.0–1000 pg/mL. A lower limit of quantification of 10.0 pg/mL was achieved with acceptable precision (<10.5%) and accuracy (5.0%). The intra- and inter-batch precision of quality control samples was <9.5%, and the accuracy ranged from −2.8% to 0.6%. The stability results showed that MA was stable in human plasma for 6 h at room temperature (25 °C), for 115 days at −20 °C, during three freeze/thaw cycles (from −20 °C to 25 °C), and in post-preparative samples for 24 h at 4 °C. The method was successfully used to characterize the pharmacokinetic profile of MA following an oral dose of 1.0 mg MA hydrate to healthy volunteers (n = 12). The proposed derivatization procedure was also extended to measure other BPs (risedronic acid and zoledronic acid) in human plasma at low pg/mL.
Co-reporter:Pan Deng, Cheng Ji, Xiaojian Dai, Dafang Zhong, Li Ding, Xiaoyan Chen
Journal of Chromatography B 2015 Volume 989() pp:71-79
Publication Date(Web):1 May 2015
DOI:10.1016/j.jchromb.2015.03.002
•Simultaneous determination of Cape and three metabolites in human plasma.•The interferences from isotopic species were chromatographically separated.•The ex vivo conversion of DFCR to DFUR in human blood was investigated.•The validated method has been successfully applied to a bioequivalence study.Capecitabine (Cape) is a prodrug that is metabolized into 5′-deoxy-5-fluorocytidine (DFCR), 5′-deoxy-5-fluorouridine (DFUR), and 5-fluorouracil (5-FU) after oral administration. A liquid chromatography–tandem mass spectrometry method for the simultaneous determination of capecitabine and its three metabolites in human plasma was developed and validated. The ex vivo conversion of DFCR to DFUR in human blood was investigated and an appropriate blood sample handling condition was recommended. Capecitabine and its metabolites were extracted from 100 μL of plasma by protein precipitation. Adequate chromatographic retention and efficient separation were achieved on an Atlantis dC18 column under gradient elution. Interferences from endogenous matrix and the naturally occurring heavy isotopic species were avoided. Detection was performed in electrospray ionization mode using a polarity-switching strategy. The method was linear in the range of 10.0–5000 ng/mL for Cape, DFCR, and DFUR, and 2.00–200 ng/mL for 5-FU. The LLOQ was established at 10.0 ng/mL for Cape, DFCR, and DFUR, and 2.00 ng/mL for 5-FU. The inter- and intra-day precisions were less than 13.5%, 11.1%, 9.7%, and 11.4%, and the accuracy was in the range of −13.2% to 1.6%, −2.4% to 2.5%, −7.1% to 8.2%, and −2.0% to 3.8% for Cape, DFCR, DFUR, and 5-FU, respectively. The matrix effect was negligible under the current conditions. The mean extraction recoveries were within 105–115%, 92.6–101%, 94.0–100%, and 85.1–99.9% for Cape, DFCR, DFUR, and 5-FU, respectively. Stability testing showed that the four analytes remained stable under all relevant analytical conditions. This method has been applied to a clinical bioequivalence study.
Co-reporter:Kan Zhong, Zhiwei Gao, Qin Li, Dafang Zhong, Xiaoyan Chen
Journal of Chromatography B 2014 Volume 961() pp:49-55
Publication Date(Web):15 June 2014
DOI:10.1016/j.jchromb.2014.04.049
•Baseline separation of morinidazole enantiomers was achieved on a cellulose chiral stationary phase column using a high proportion of organic phase.•d4-Morinidazole was synthesized as the internal standard to compensate for matrix effects and variables during sample preparation.•The analysis time was short by using a flow rate gradient.•The method shows advantages of high selectivity and reproducibility.•The method was successfully applied to clinical stereoselective pharmacokinetics study of morinidazole.Morinidazole is a novel 5-nitroimidazole derivative used for the treatment of amoebiasis, trichomoniasis, and anaerobic bacterial infections. Morinidazole possesses a chiral carbon and is clinically administered as a racemate. In the present study, an enantioselective and sensitive liquid chromatography–tandem mass spectrometry method of determining morinidazole enantiomers in human plasma was developed and validated to characterize the stereoselective pharmacokinetics. Plasma samples were processed by liquid–liquid extraction using tert-butyl methyl ether. Chiral separation was optimized within 8.5 min on a cellulose column using an isocratic mobile phase of methanol/water (80:20, v/v). Detection was using mass spectrometry in multiple reaction monitoring mode, using the transitions of m/z 271 → 144 for morinidazole enantiomers, and m/z 275 → 148 for d4-morinidazole (internal standard). The calibration curves were linear over 5.00–6000 ng/mL for each enantiomer. The lower limit of quantification for each enantiomer was established at 5.00 ng/mL. Intra- and inter-day precisions were less than 6.4% for each enantiomer in terms of relative standard deviation, and accuracies were between −2.5% and 6.4% in terms of relative error for each enantiomer. No chiral inversion was observed during sample storage, preparation procedure and analysis. Major glucuronide and sulfate conjugates were not observed to interfere with the determination of morinidazole enantiomers. The method was applied to study the stereoselective pharmacokinetics of morinidazole in humans. Moderate stereoselectivity was observed in healthy subjects and patients with severe renal impairment.
Co-reporter:Cen Xie;Jialan Zhou;Zitao Guo;Xingxing Diao;Zhiwei Gao;Dafang Zhong;Haoyuan Jiang;Lijia Zhang
British Journal of Pharmacology 2013 Volume 168( Issue 7) pp:1687-1706
Publication Date(Web):
DOI:10.1111/bph.12047
Background and Purpose
Famitinib is a novel multi-targeted receptor tyrosine kinase inhibitor under development for cancer treatment. This study aims to characterize the metabolic and bioactivation pathways of famitinib.
Experimental Approach
The metabolites in human plasma, urine and feces were identified via ultra-high performance liquid chromatography-quadrupole-time of flight-mass spectrometry and confirmed using synthetic standards. Biotransformation and bioactivation mechanisms were investigated using microsomes, recombinant metabolic enzymes and hepatocytes.
Key Results
Famitinib was extensively metabolized after repeated oral administrations. Unchanged famitinib was the major circulating material, followed by N-desethylfaminitib (M3), whose steady-state exposure represented 7.2 to 7.5% that of the parent drug. Metabolites in the excreta were mainly from oxidative deamination (M1), N-desethylation (M3), oxidative defluorination (M7), indolylidene hydroxylation (M9-1 and M9-5) and secondary phase-II conjugations. CYP3A4/5 was the major contributor to M3 formation, CYP3A4/5 and aldehyde dehydrogenase to M1 formation and CYP1A1/2 to M7, M9-1 and M9-5 formations. Minor cysteine conjugates were observed in the plasma, urine and feces, implying the formation of reactive intermediate(s). In vitro microsomal studies proved that famitinib was bioactivated through epoxidation at indolylidene by CYP1A1/2 and spontaneously defluorinated rearrangement to afford a quinone-imine species. A correlation between famitinib hepatotoxicity and its bioactivation was observed in the primary human hepatocytes.
Conclusion and Implications
Famitinib is well absorbed and extensively metabolized in cancer patients. Multiple enzymes, mainly CYP3A4/5 and CYP1A1/2, are involved in famitinib metabolic clearance. The quinone-imine intermediate formed through bioactivation may be associated with famitinib hepatotoxicity. Co-administered CYP1A1/2 inducers or inhibitors may potentiate or suppress its hepatotoxicity.
Co-reporter:Xingxing Diao, Zhiyu Ma, Haidong Wang, Dafang Zhong, Yifan Zhang, Jing Jin, Yaxin Fan, Xiaoyan Chen
Journal of Pharmaceutical and Biomedical Analysis 2013 Volumes 78–79() pp:19-26
Publication Date(Web):5 May 2013
DOI:10.1016/j.jpba.2013.01.033
3-n-Butylphthalide (NBP) is a cardiovascular drug widely used in China for the treatment of cerebral ischemic stroke. Our previous study showed that NBP underwent extensive metabolism in humans and that 10-keto-NBP (M2), 3-hydroxy-NBP (M3-1), 10-hydroxy-NBP (M3-2) and NBP-11-oic acid (M5-2) were the major circulating metabolites. A better understanding of the plasma exposures of NBP and its four major metabolites is crucial to supporting the safety evaluation, good clinic practice and discovery of new antistroke drugs. Herein, a liquid chromatography–tandem mass spectrometry method was developed and validated for the simultaneous determination of NBP, M2, M3-1, M3-2, and M5-2 in human plasma. To improve assay sensitivity and achieve simultaneous analysis, M3-1 and M5-2 were monitored in (−)ESI (electrospray) mode within the first 3.5 min and NBP, M2, and M3-2 thereafter in (+)ESI mode. Deuterated internal standards for all analytes were synthesized to compensate for the impact of matrix effects. Based on the vast differences in physicochemical properties among the five analytes and the necessary baseline separation of two isomers (i.e., M3-1 and M3-2), gradient elution comprising a mobile phase of methanol–acetonitrile–5 mM ammonium acetate was used after methanol-induced protein precipitation of plasma samples. The developed method was linear in the concentration range of 3.00–800 ng/ml for NBP and M2, and 3.00–2400 ng/ml for M3-1, M3-2, and M5-2. The lower limit of quantitation was 3.00 ng/ml for each analyte. The intra- and inter-day accuracy and precision were within acceptable limits (±15%) at all concentrations. The method was successfully applied to characterize the pharmacokinetic profiles of NBP and its major metabolites following a single oral administration of 200 mg NBP to healthy volunteers.Graphical abstractHighlights► Simultaneous determination of NBP and its four major metabolites in human plasma. ► Synthetic deuterated ISs were utilized to compensate for matrix effects. ► (−)ESI and (+)ESI switch was complementary to achieve high sensitivity. ► Mixture of methanol and acetonitrile facilitated separation of isomers.
Co-reporter:Yong Yang, Jinghua Yu, Youming Lu, Yu Xia, Dafang Zhong, Xiaoyan Chen
Journal of Chromatography B 2013 Volumes 915–916() pp:1-7
Publication Date(Web):1 February 2013
DOI:10.1016/j.jchromb.2012.12.007
Sodium picosulfate (PICO-Na) is a member of the polyphenolic group of stimulant laxatives. Its major metabolites in humans are its active aglycone BHPM (bis-(p-hydroxyphenyl)-pyridyl-2-methane), the monoglucuronide (M1) and the monosulfate (M2) of BHPM. A sensitive, specific and rapid liquid chromatography–tandem mass spectrometry method was established and validated for the simultaneous determination of picosulfate (PICO) and its three major metabolites in human plasma to investigate the pharmacokinetics of PICO and its major metabolites. Following protein precipitation with acetonitrile, chromatographic separation was achieved on a Luna 5u C18(2) column using gradient elution starting with 10% of 10 mM ammonium acetate followed by increasing percentages of acetonitrile to eliminate interferences due to in-source conversion of the conjugated metabolites. Detection was performed on a tandem mass spectrometer equipped with an electrospray ionization source operated in the positive mode, using the transitions of m/z 438.1 → m/z 278.1 for PICO, m/z 278.1 → m/z 184.2 for BHPM, m/z 454.1 → m/z 184.2 for M1, and m/z 358.1 → m/z 184.2 summed with m/z 358.1 → m/z 278.1 for M2. Deuterium labeled compounds of the analytes were used as the internal standard, two of which, M1-d12 and M2-d12, were synthesized in-house. The method was validated in concentration ranges of 0.150–40.0 ng/mL for PICO and M2, 0.600–160 ng/mL for BHPM, and 0.045–12.0 ng/mL for M1 with acceptable accuracy and precision. The method was successfully applied to characterize the pharmacokinetic profiles of PICO and its metabolites in healthy volunteers after a single oral administration of 5 mg PICO-Na.Highlights► An LC/MS/MS method for simultaneous assay PICO and its metabolites in human plasma. ► In-source conversion interferences of conjugated compounds were avoided by chromatography separation. ► Reference standards for M1, M2 and their deuterium labeled ISs were in-house synthesized. ► Deuterium labeled ISs were used to improve the reproducibility. ► The pharmacokinetic profiles of PICO, M1, and M2 in humans were firstly characterized.
Co-reporter:Meixia Chen, Yu Xia, Zhiyu Ma, Liang Li, Dafang Zhong, Xiaoyan Chen
Journal of Chromatography B 2012 Volume 906() pp:85-90
Publication Date(Web):1 October 2012
DOI:10.1016/j.jchromb.2012.08.019
Pantoprazole (PAN), a selective proton pump inhibitor, is used clinically as a racemic mixture for the treatment of acid-related gastrointestinal disorders. To investigate its stereoselective pharmacokinetics, a chiral liquid chromatography–tandem mass spectrometry method was developed and validated to determine the pantoprazole enantiomers in dog plasma. After liquid–liquid extraction, a baseline resolution of enantiomers was achieved on an ovomucoid column using the mobile phase of methanol:acetonitrile:10 mM ammonium formate (pH 7) (10.4:2.6:87, v/v/v) at 30 °C within 10 min. Stable isotopically labeled (+)-d3-pantoprazole and (−)-d3-pantoprazole were used as internal standards. Acquisition of mass spectrometric data was performed in multiple reaction monitoring mode via positive atmospheric pressure chemical ionization. The method was linear in the concentration range of 20.0–10,000 ng/mL for each enantiomer using 25 μL of dog plasma. The lower limit of quantification (LLOQ) for each enantiomer was 20.0 ng/mL. Intra- and inter-day precision ranged from 3.2% to 10.3% for (+)-pantoprazole and 3.7–10.0% for (−)-pantoprazole. Accuracy varied from −1.4% to −0.2% for (+)-pantoprazole and −1.6% to 0.8% for (−)-pantoprazole. The validated method was applied successfully for stereoselective pharmacokinetic studies of racemic pantoprazole.Highlights► A chiral LC–MS/MS method was validated to quantify pantoprazole enantiomers. ► Separation was performed on an ovomucoid protein column using MS compatible mobile phases. ► Baseline resolution within 10 min leads to a reduction in the overall analysis time.
Co-reporter:Ruina Gao, Dafang Zhong, Ke Liu, Yu Xia, Rongwei Shi, Hua Li, Xiaoyan Chen
Journal of Chromatography B 2012 Volume 908() pp:52-58
Publication Date(Web):1 November 2012
DOI:10.1016/j.jchromb.2012.09.017
Morinidazole is a new third-generation 5-nitroimidazole antimicrobial drug. To investigate the pharmacokinetic profiles of morinidazole and its major metabolites in humans, a liquid chromatography–tandem mass spectrometry method was developed and validated for simultaneous determination of morinidazole, its N-oxide metabolite (M4-1), a sulfate conjugate (M7), and two diastereoisomeric N+-glucuronides (M8-1 and M8-2) in human plasma. A simple acetonitrile-induced protein precipitation was employed to extract five analytes and internal standard metronidazole from 50 μL human plasma. To avoid the interference from the in-source dissociation of the sulfate and achieve the baseline-separation of diastereoisomeric N+-glucuronides, all the analytes were separated from each other with the mobile phase consisting of 10 mM ammonium formate and acetonitrile using gradient elution on a Hydro-RP C18 column (50 mm × 2 mm, 4 μm) with a total run time of 5 min. The API 4000 triple quadrupole mass spectrometer was operated under the multiple reaction-monitoring mode using the electrospray ionization technique. The developed method was linear in the concentration ranges of 10.0–12,000 ng/mL for morinidazole, 1.00–200 ng/mL for M4-1, 2.50–500 ng/mL for M7, 3.00–600 ng/mL for M8-1, and 10.0–3000 ng/mL for M8-2. The intra- and inter-day precisions for each analyte met the accepted value. Results of the stability of morinidazole and its metabolites in human plasma were also presented. The method was successfully applied to the clinical pharmacokinetic studies of morinidazole injection in healthy subjects, patients with moderate hepatic insufficiency, and patients with severe renal insufficiency, respectively.Highlights► Simultaneously determine morinidazole and its four metabolites in human plasma. ► Gradient elution was used to obtain the resolution of two N+-glucuronide isomers. ► The potential interference from the in-source dissociation of the conjugates was avoided. ► The method was applied to clinical pharmacokinetic studies of morinidazole injection.
Co-reporter:Yong Yang, Ke Liu, Dafang Zhong, Xiaoyan Chen
Journal of Chromatography B 2012 Volumes 895–896() pp:25-30
Publication Date(Web):1 May 2012
DOI:10.1016/j.jchromb.2012.03.008
Flumatinib is an antineoplastic tyrosine kinase inhibitor used for the treatment of chronic myelogenous leukemia (CML). Its major metabolites in the circulation are N-desmethyl flumatinib (M1) and amide hydrolysis product (M3). To investigate the pharmacokinetics of flumatinib in CML patients, a simple, specific and rapid liquid chromatography–tandem mass spectrometry (LC–MS/MS) method was developed and validated for the simultaneous determination of flumatinib and its two major metabolites in patient plasma. After a simple, one-step protein precipitation with methanol, flumatinib, its two metabolites, and internal standard (HHGV-E) were separated on a C18 column using an isocratic mobile phase of methanol:5 mM ammonium acetate:formic acid (60:40:0.4, v/v/v). A total chromatographic run time of 4.2 min was achieved. The detection was performed in multiple reaction monitoring mode, using the transitions of m/z 563 → m/z 463 for flumatinib, m/z 549 → m/z 463 for M1, m/z 303 → m/z 175 for M3, and m/z 529 → m/z 429 for HHGV-E. The method was linear over the concentration ranges of 0.400–400 ng/mL for flumatinib, 0.100–100 ng/mL for M1, and 0.200–200 ng/mL for M3, using only 50 μL of plasma. The intra- and inter-day precisions were less than 8.5% for flumatinib, 9.8% for M1, and 10.6% for M3 in terms of the relative standard deviation. The accuracy was within ±2.2% for flumatinib, ±6.0% for M1, and ±9.9% for M3 in terms of relative error. The validated method was successfully applied to clinical pharmacokinetic studies of flumatinib mesylate in CML patients following oral administration at all dosage regimens.Highlights► Simultaneously determine flumatinib and its metabolites in CML patient plasma. ► Flumatinib and its metabolites have great differences in physicochemical properties. ► Each analyte was retained on the C18 column by using strong acidic mobile phase. ► A simple one-step protein precipitation increased throughput and efficiency. ► The method was applied to clinical studies of flumatinib mesylate in CML patients.
Co-reporter:Cen Xie, Kate Yu, Dafang Zhong, Tao Yuan, Fei Ye, Joseph Andy Jarrell, Alan Millar, and Xiaoyan Chen
Journal of Agricultural and Food Chemistry 2011 Volume 59(Issue 20) pp:11078-11087
Publication Date(Web):September 25, 2011
DOI:10.1021/jf203104k
Ultraperformance liquid chromatography coupled with hybrid quadrupole/ion mobility/orthogonal acceleration time-of-flight (oa-TOF) mass spectrometry (UPLC-IM-MS) was used to study the isomeric transformations of trans-5-caffeoylquinic acid, an extremely active compound present in multiple vegetables, fruits, and beverages. The UPLC/oa-TOF MS results proved that in phosphate buffer (pH 7.4), plasma, or urine sample, trans-5-caffeoylquinic acid first isomerizes to trans-4-caffeoylquinic acid and then to trans-3-caffeoylquinic acid by intramolecular acyl migration. When exposed to UV light, trans-3-, -4-, and -5-caffeoylquinic acids undergo cis/trans isomerization to form cis isomers. The isomerization was solely dependent on the pH of the matrix, as well as the incubation temperature, and was independent of metabolic enzymes. UPLC-IM-MS results revealed that a reversible cis/trans isomerization of caffeoylquinic acids could also be induced by the electric field in an electrospray source. Thus, understanding the possible role of electric field-induced isomerization of caffeoylquinic acids may help lessen the confusion between gas phase phenomena and liquid state chemistry when applying IM-MS analysis. The comprehensive understanding of caffeoylquinic acid isomerization transformations is crucial for the appropriate handling of samples and interpretation of experimental data.
Co-reporter:Hua Jin, Liang Li, Dafang Zhong, Jia Liu, Xiaoyan Chen, and Jiang Zheng
Chemical Research in Toxicology 2011 Volume 24(Issue 12) pp:2142
Publication Date(Web):October 12, 2011
DOI:10.1021/tx200290s
Tetrandrine, a bisbenzylisoquinoline alkaloid, has demonstrated promising pharmacologic activities. The alkaloid has a great potential for clinical use, so a careful, thorough toxicity evaluation of the alkaloid is required. In the present study, 24 h acute toxicity of tetrandrine was evaluated in CD-1 mice. Single intraperitoneal doses of tetrandrine at 150 mg (0.24 mmol)/kg were found to cause alveolar hemorrhage and over 3-fold elevation of lactate dehydrogenase activity in bronchoalveolar lavage fluids. Ethidium-based staining showed loss of membrane integrity in significant numbers of cells in the lungs of the animals treated with the same doses of tetrandrine. As much as 60% reduction in cell viability was observed after 24 h of exposure to tetrandrine at 40 μM in human lung cell lines NL-20 and WI-38. Ketoconazole, an inhibitor of P450 3A, showed a protective effect on the pulmonary injury in mice given tetrandrine. A glutathione (GSH) conjugate derived from O-demethylated tetrandrine was detected in incubations of tetrandrine with NADPH- and GSH-supplemented human liver and mouse lung microsomes. The electrophilic metabolite trapped by GSH is considered to be a quinone methide derivative. The formation of the metabolite reactive to GSH was found to require the presence of NADPH. The coincubation of ketoconazole suppressed the generation of the GSH conjugate. Tetrandrine was incubated with a selection of recombinant human cytochrome P450 enzymes, and only P450s 3A4 and 3A5 were responsible for the production of the reactive metabolite. The results implicate a possible correlation between the formation of the quinone methide metabolite of tetrandrine and the pulmonary toxicity induced by tetrandrine.
Co-reporter:Cen Xie, Shilei Yang, Dafang Zhong, Xiaojian Dai, Xiaoyan Chen
Journal of Chromatography B 2011 Volume 879(Issue 28) pp:3071-3075
Publication Date(Web):15 October 2011
DOI:10.1016/j.jchromb.2011.09.004
Dronedarone is a derivative of amiodarone – a popular antiarrhythmic drug. It was developed to overcome the limiting iodine-associated toxicities of amiodarone. Debutyldronedarone is a major circulating active metabolite of dronedarone in humans. To investigate the pharmacokinetics of dronedarone, a rapid, simple, and sensitive liquid chromatography–tandem mass spectrometry (LC–MS/MS) method was developed and validated to simultaneously determine dronedarone and debutyldronedarone in human plasma using amiodarone as internal standard (IS). Acetonitrile with IS was used to precipitate proteins from a 50-μL aliquot of plasma. Effective chromatographic separation was performed on a CAPCELL PAK C18 MG (100 mm × 4.6 mm, 5 μm) column with gradient elution (5 mmol/L ammonium acetate–acetonitrile, with each phase containing 0.2% acetic acid) at a flow rate of 0.7 mL/min. Complete separation was achieved within 5.5 min. Detection was carried out on an tandem mass spectrometer in multiple reaction monitoring mode using a positive atmospheric pressure chemical ionization interface. A lower limit of quantification of 0.200 ng/mL was achieved for both dronedarone and debutyldronedarone, with acceptable precision and accuracy. The linear range of the method was from 0.200 to 200 ng/mL for each analyte. Intra- and inter-day precisions were lower than 7.2% in relation to relative standard deviation, while accuracy was within ±5.1% in terms of relative error for analytes. Our findings demonstrate the successful application of the validated LC–MS/MS method to a pharmacokinetic study after a single oral administration of 400 mg dronedarone to six healthy volunteers.Highlights► An LC-MS/MS method was validated to determine dronedarone and debutyldronedarone in human plasma. ► The method has an LLOQ of 0.200 ng/mL for both analytes. ► Sample preparation was simple and quick with acetonitrile protein precipitation. ► The method was applied to a pharmacokinetic study of dronedarone in humans.
Co-reporter:Pan Deng, Dafang Zhong, Fajun Nan, Sheng Liu, Dan Li, Tao Yuan, Xiaoyan Chen, and Jiang Zheng
Chemical Research in Toxicology 2010 Volume 23(Issue 10) pp:1617
Publication Date(Web):September 15, 2010
DOI:10.1021/tx100223h
4-Nonylphenol (4-NP) is a well-known toxic environmental contaminant. The major objective of the present study was to identify reactive metabolites of 4-NP. Following incubations of 4-NP with NADPH- and GSH-supplemented human liver microsomes, 6 GSH conjugates, along with 19 oxidized metabolites, were detected by UPLC/Q-TOF mass spectrometry utilizing the mass defect filter method. Several authentic key metabolite standards were chemically synthesized for structural identification. Three GSH conjugates were found to derive from quinone methide intermediates, and the other three resulted from ortho-benzoquinone intermediates. Conjugation of the quinone methides with GSH produced benzylic-orientated GSH conjugates by 1,6-addition, while the reaction of the ortho-benzoquinone intermediates offered aromatic-orientated GSH conjugates. The conversion of 4-NP to the quinone methides and ortho-hydroquinones required cytochromes P450, specifically CYPs1A2, 2C19, 2D6, 2E1, and 3A4, while the oxidation of ortho-benzohydroquinones to the corresponding benzoquinones was apparently independent of microsomal enzymes. The ortho-benzoquinone derived from 4-NP was isomerized to the corresponding hydroxyquinone methide, and the former dominated the latter at a rate of approximately 20:1. The findings of the quinone methide and benzoquinone metabolites intensified the concern on the impact of 4-NP exposure on human health.
Co-reporter:Ke Liu, Dafang Zhong, Huiyu Zou, Xiaoyan Chen
Journal of Pharmaceutical and Biomedical Analysis 2010 52(4) pp: 550-556
Publication Date(Web):
DOI:10.1016/j.jpba.2010.01.026
Co-reporter:Ke Liu, Dafang Zhong, Xiaoyan Chen
Journal of Chromatography B 2010 Volume 878(Issue 26) pp:2415-2420
Publication Date(Web):15 September 2010
DOI:10.1016/j.jchromb.2010.07.027
An enantioselective and sensitive method was developed and validated for determination of doxazosin enantiomers in human plasma by liquid chromatography–tandem mass spectrometry. The enantiomers of doxazosin were extracted from plasma using ethyl ether/dichloromethane (3/2, v/v) under alkaline conditions. Baseline chiral separation was obtained within 9 min on an ovomucoid column using an isocratic mobile phase of methanol/5 mM ammonium acetate/formic acid (20/80/0.016, v/v/v) at a flow rate of 0.60 mL/min. Acquisition of mass spectrometric data was performed in multiple reaction monitoring mode, using the transitions of m/z 452 → 344 for doxazosin enantiomers, and m/z 384 → 247 for prazosin (internal standard). The method was linear in the concentration range of 0.100–50.0 ng/mL for each enantiomer using 200 μL of plasma. The lower limit of quantification (LLOQ) for each enantiomer was 0.100 ng/mL. The intra- and inter-assay precision was 5.0–11.1% and 5.7–7.6% for R-(−)-doxazosin and S-(+)-doxazosin, respectively. The accuracy was 97.4–99.5% for R-(−)-doxazosin and 96.8–102.8% for S-(+)-doxazosin. No chiral inversion was observed during the plasma storage, preparation and analysis. The method proved adequate for enantioselective pharmacokinetic studies of doxazosin after oral administration of therapeutic doses of racemic doxazosin.
Co-reporter:Yuya Wang, Dafang Zhong, Xiaoyan Chen and Jiang Zheng
Chemical Research in Toxicology 2009 Volume 22(Issue 5) pp:824
Publication Date(Web):April 9, 2009
DOI:10.1021/tx800397e
Dauricine is one type of the bisbenzyltetrahydroisoquinoline alkaloid derivative with antiarrhythmic effects. Severe liver toxicity was observed in experimental animals treated with analogues of dauricine, which may be caused by covalent binding of reactive metabolite(s) to critical macromolecules in tissues. The study described herein aimed at characterizing pathways of dauricine bioactivation and the CYP enzyme involved. In incubations of dauricine with NADPH- and GSH-supplemented human liver microsomes, four GSH conjugates with [M + H]+ ions at m/z 930, 916, 916, and 902, respectively, were detected by liquid chromatography-ion trap mass spectrometry. The structures of the four metabolites were determined to be GSH conjugates of dauricine, 2-N-demethyl dauricine, 2′-N-demethyl dauricine, and N-demethyl-O-demethyl dauricine. GSH conjugation took place with a strong preference at C-17, suggesting that the phenol moiety of dauricine and its metabolites underwent oxidation to quinone methide intermediates. The formation of the GSH conjugates was found to require the presence of NADPH. To identify the CYP isoforms that are responsible for bioactivation, dauricine was also incubated with recombinant human CYP450 enzymes. The formation of GSH was only observed with the incubation of CYP3A4. In addition, the level of these GSH conjugates in human microsomes was reduced upon the addition of a CYP3A4 inhibitor ketoconazole. The same GSH conjugates were also observed in rat bile following a single oral dose of 40 mg/kg dauricine. These studies suggest that the CYP3A4 mediated quinone methide formation was associated with dauricine bioactivation.
Co-reporter:Pan Deng, Dafang Zhong, Xiaoyan Chen
Journal of Pharmaceutical and Biomedical Analysis 2009 49(3) pp: 848-852
Publication Date(Web):
DOI:10.1016/j.jpba.2008.12.038
Co-reporter:Ke Liu, Xiaojian Dai, Dafang Zhong, Pan Deng, Jinfei Ma, Xiaoyan Chen
Journal of Chromatography B 2009 Volume 877(Issue 10) pp:902-910
Publication Date(Web):1 April 2009
DOI:10.1016/j.jchromb.2009.02.046
A selective and rapid method was developed and validated for determination of 6R-leucovorin (LV), 6S-leucovorin and 5-methyltetrahydrofolate (5-MeTHF) in human plasma using stereoselective liquid chromatography–tandem mass spectrometry. All analytes and the internal standard were extracted from plasma by solid phase extraction using Oasis® HLB C18 cartridges. A macrocyclic glycopeptide teicoplanin column was used for chiral separation of LV and 5-MeTHF isomers with NH4TFA or NH4OAc in methanol as mobile phase. Detection was performed on an API 4000 tandem mass spectrometer with positive electrospray ionization in multiple reaction monitoring mode. The calibration curves were linear in the range of 0.050–20.0 μg/mL for 6R-LV and 6S-LV, and 0.025–10.0 μg/mL for 5-MeTHF. The intra- and inter-assay precision was 3.6–13.2%, 3.4–12.9% and 5.3–9.3% for 6R-LV, 6S-LV and 5-MeTHF, respectively. The accuracy was 99.4–102.4%, 95.3–96.8% and 93.0–110% for 6R-LV, 6S-LV and 5-MeTHF, respectively. The lower limit of quantification (LLOQ) was 0.050 μg/mL for each LV isomer and 0.025 μg/mL for 5-MeTHF. The method was successfully applied to a comparative pharmacokinetic study between leucovorin calcium and levoleucovorin calcium in 10 volunteers. No significant differences between levoleucovorin and leucovorin in pharmacokinetic parameters of 6S-LV and 5-MeTHF were found in volunteers.
Co-reporter:Huaicheng Zhang, Dafang Zhong, Zhenzhong Zhang, Xiaojian Dai, Xiaoyan Chen
Journal of Chromatography B 2009 Volume 877(Issue 27) pp:3221-3225
Publication Date(Web):1 October 2009
DOI:10.1016/j.jchromb.2009.06.005
A sensitive and rapid liquid chromatography/tandem mass spectrometric (LC/MS/MS) method was developed and validated for the determination of deserpidine in human plasma. The plasma samples were prepared using liquid–liquid extraction (LLE) with ethyl ether–dichloromethane (3:2, v/v). Chromatographic separation was accomplished on an Ultimate XB-C18 column. The mobile phase consisted of methanol–5 mM ammonium acetate–formic acid (72:28:0.036, v/v/v). Detection of deserpidine and the internal standard tropisetron was achieved by tandem mass spectrometry with an electrospray ionization interface in positive ion mode. The lower limit of quantification was 4.0 pg/ml. The linear range of the method was from 4.0 to 2000 pg/ml. The intra- and inter-day precisions were lower than 14.7% in terms of relative standard deviation (RSD), and the accuracy was within ±8.7% in terms of relative error (RE). This validated method was successfully applied for the evaluation of pharmacokinetics of deserpidine after a single oral administration dose of 0.25 mg deserpidine to 22 healthy volunteers.
Co-reporter:Jun Gao, Dafang Zhong, Xiaotao Duan, Xiaoyan Chen
Journal of Chromatography B 2007 Volume 856(1–2) pp:35-40
Publication Date(Web):1 September 2007
DOI:10.1016/j.jchromb.2007.05.012
A sensitive liquid chromatography/tandem mass spectrometric (LC–MS/MS) method was developed and validated for the determination of rosuvastatin in human plasma. The plasma samples were prepared using liquid–liquid extraction with ethyl ether. Chromatographic separation was accomplished on a Zorbax XDB-C18 (150 mm × 4.6 mm i.d., 5 μm) column. The mobile phase consisted of methanol–water (75:25, v/v, adjusted to pH 6 by aqueous ammonia). Detection of rosuvastatin and the internal standard (IS) hydrochlorothiazide was achieved by ESI MS/MS in the negative ion mode. The lower limit of quantification was 0.020 ng/ml by using 200 μl aliquots of plasma. The linear range of the method was from 0.020 to 60.0 ng/ml. The intra- and inter-day precisions were lower than 8.5% in terms of relative standard deviation (RSD), and the accuracy was within −0.3 to 1.9% in terms of relative error (RE). Compared with the existing methods, the validated method offered increased sensitivity. The method was successfully applied for the evaluation of pharmacokinetics of rosuvastatin after single oral doses of 5, 10 and 20 mg rosuvastatin to 10 healthy volunteers.
Co-reporter:Sheng Zhong, Dafang Zhong, Xiaoyan Chen
Journal of Chromatography B 2007 Volume 854(1–2) pp:291-298
Publication Date(Web):1 July 2007
DOI:10.1016/j.jchromb.2007.04.043
Glucosamine is an amino monosaccharide reagent. It is difficult to assay using typical reversed-phase column due to the early elution, by optimizing the chromatographic conditions, especially the analytical column and the mobile phase composition, an improved analytical method was developed and validated, which offers rapid, sensitive and specific determination of glucosamine in human plasma. Following protein precipitation, the analyte and internal standard (valibose) were separated using an isocratic mobile phase on an Inertsil CN-3 column and detected by mass spectrometry in the multiple reaction monitoring mode using the respective precursor to product ion combinations of m/z 180/72 for glucosamine and m/z 252/198 for valibose. The chromatographic time was just 4.2 min for each sample, which made it possible to analyze more than 120 human plasma samples per day. The method exhibited a linear dynamic range of 4.00–4000 ng/mL for glucosamine in human plasma. The lower limit of quantification (LLOQ) was 4.00 ng/mL with a relative standard deviation of less than 10.9%. Acceptable precision and accuracy were obtained for the plasma concentrations over the standard curve range. By monitoring the two different MRM transitions, it was proved that no endogenous glucosamine was found in human plasma. The validated method has been successfully used to analyze human plasma samples for application in a bioequivalence study.

![N-[4-(1-Cyanocyclopentyl)phenyl]-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide methanesulfonate](http://img.cochemist.com/ccimg/1218800/1218779-75-9.png)
![N-[4-(1-Cyanocyclopentyl)phenyl]-2-[(4-pyridinylmethyl)amino]-3-pyridinecarboxamide methanesulfonate](http://img.cochemist.com/ccimg/1218800/1218779-75-9_b.png)
![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-5-(4-fluorophenyl)-1,4-dihydro-4-oxo-](/data/chemimg/390900/1174046-72-0.png)
![3-Pyridinecarboxamide, N-[4-[(2-amino-3-chloro-4-pyridinyl)oxy]-3-fluorophenyl]-5-(4-fluorophenyl)-1,4-dihydro-4-oxo-](/data/chemimg/390900/1174046-72-0_b.png)
![Benzonitrile, 3-[1,6-dihydro-1-[[3-[5-[(1-methyl-4-piperidinyl)methoxy]-2-pyrimidinyl]phenyl]methyl]-6-oxo-3-pyridazinyl]-](/data/chemimg/345600/1100598-32-0.png)
![Benzonitrile, 3-[1,6-dihydro-1-[[3-[5-[(1-methyl-4-piperidinyl)methoxy]-2-pyrimidinyl]phenyl]methyl]-6-oxo-3-pyridazinyl]-](/data/chemimg/345600/1100598-32-0_b.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8.png)
![Benzamide, 2-fluoro-N-methyl-4-[7-(6-quinolinylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]-](/data/chemimg/315100/1029712-80-8_b.png)
![2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyr azol-1-yl)ethanol](http://img.cochemist.com/ccimg/957000/956905-27-4.png)
![2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyr azol-1-yl)ethanol](http://img.cochemist.com/ccimg/957000/956905-27-4_b.png)





![1,1-Cyclopropanedicarboxamide, N-[2-fluoro-4-[[2-[[[4-(4-methyl-1-piperazinyl)-1-piperidinyl]carbonyl]amino]-4-pyridinyl]oxy]phenyl]-N'-(4-fluorophenyl)-](/data/chemimg/2243600/928037-13-2.png)
![1,1-Cyclopropanedicarboxamide, N-[2-fluoro-4-[[2-[[[4-(4-methyl-1-piperazinyl)-1-piperidinyl]carbonyl]amino]-4-pyridinyl]oxy]phenyl]-N'-(4-fluorophenyl)-](/data/chemimg/2243600/928037-13-2_b.png)
![1H-Pyrazole-4-carboxamide, 2,3-dihydro-1-(2-hydroxy-2-methylpropyl)-N-[5-[(7-methoxy-4-quinolinyl)oxy]-2-pyridinyl]-5-methyl-3-oxo-2-phenyl-](/data/chemimg/1589500/913376-83-7.png)
![1H-Pyrazole-4-carboxamide, 2,3-dihydro-1-(2-hydroxy-2-methylpropyl)-N-[5-[(7-methoxy-4-quinolinyl)oxy]-2-pyridinyl]-5-methyl-3-oxo-2-phenyl-](/data/chemimg/1589500/913376-83-7_b.png)
![PIPERIDINE, 3-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/876200/876147-50-1.png)
![PIPERIDINE, 3-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/876200/876147-50-1_b.png)
![Benzeneacetamide, N-[[[3-fluoro-4-[[2-(1-methyl-1H-imidazol-4-yl)thieno[3,2-b]pyridin-7-yl]oxy]phenyl]amino]thioxomethyl]-](/data/chemimg/1587400/875337-44-3.png)
![Benzeneacetamide, N-[[[3-fluoro-4-[[2-(1-methyl-1H-imidazol-4-yl)thieno[3,2-b]pyridin-7-yl]oxy]phenyl]amino]thioxomethyl]-](/data/chemimg/1587400/875337-44-3_b.png)

















![Benzenesulfonamide,3-(4,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-](http://img.cochemist.com/ccimg/319500/319491-68-4.png)
![Benzenesulfonamide,3-(4,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-propoxy-](http://img.cochemist.com/ccimg/319500/319491-68-4_b.png)
![calcium,(3S,5S)-7-[2-(4-fluorophenyl)-4-[(2-hydroxyphenyl)carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate](http://img.cochemist.com/ccimg/266000/265989-46-6.png)
![calcium,(3S,5S)-7-[2-(4-fluorophenyl)-4-[(2-hydroxyphenyl)carbamoyl]-3-phenyl-5-propan-2-ylpyrrol-1-yl]-3,5-dihydroxyheptanoate](http://img.cochemist.com/ccimg/266000/265989-46-6_b.png)
![2-Oxa-6-azaspiro[3.4]octane](http://img.cochemist.com/ccimg/220300/220290-68-6.png)
![2-Oxa-6-azaspiro[3.4]octane](http://img.cochemist.com/ccimg/220300/220290-68-6_b.png)
![(1-Hydroxy-2-(imidazo[1,2-a]pyridin-3-yl)ethane-1,1-diyl)diphosphonic acid](http://img.cochemist.com/ccimg/180100/180064-38-4.png)
![(1-Hydroxy-2-(imidazo[1,2-a]pyridin-3-yl)ethane-1,1-diyl)diphosphonic acid](http://img.cochemist.com/ccimg/180100/180064-38-4_b.png)
![Pyrrolidine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/159400/159330-31-1.png)
![Pyrrolidine, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-, (3R)-](http://img.cochemist.com/ccimg/159400/159330-31-1_b.png)






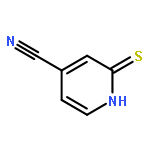
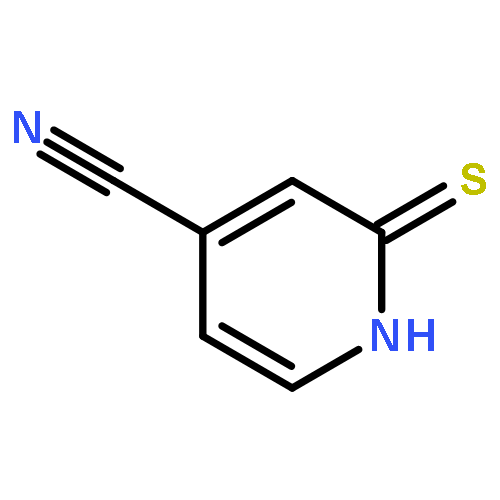
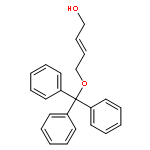

![Propanoic acid, 2-[[[N-(Z)]-[1-(2-amino-4-thiazolyl)-2-[[(3S)-1-[[[[3-(1,4-dihydro-5-hydroxy-4-oxo-2-pyridinyl)-4,5-dihydro-4-methyl-5-oxo-1H-1,2,4-triazol-](/data/chemimg/402000/123444-35-9.png)
![Propanoic acid, 2-[[[N-(Z)]-[1-(2-amino-4-thiazolyl)-2-[[(3S)-1-[[[[3-(1,4-dihydro-5-hydroxy-4-oxo-2-pyridinyl)-4,5-dihydro-4-methyl-5-oxo-1H-1,2,4-triazol-](/data/chemimg/402000/123444-35-9_b.png)
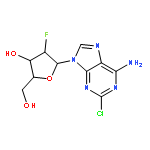


![Benzonitrile, 4-[(6,7-dihydro-7-hydroxy-5H-cyclopentapyrimidin-4-yl)amino]-](/data/chemimg/315300/105365-76-2.png)
![Benzonitrile, 4-[(6,7-dihydro-7-hydroxy-5H-cyclopentapyrimidin-4-yl)amino]-](/data/chemimg/315300/105365-76-2_b.png)
![Acetic acid,2-[[(Z)-[1-(2-amino-4-thiazolyl)-2-[[(3S)-2,2-dimethyl-4-oxo-1-(sulfooxy)-3-azetidinyl]amino]-2-oxoethylidene]amino]oxy]-](http://img.cochemist.com/ccimg/102600/102507-71-1.png)
![Acetic acid,2-[[(Z)-[1-(2-amino-4-thiazolyl)-2-[[(3S)-2,2-dimethyl-4-oxo-1-(sulfooxy)-3-azetidinyl]amino]-2-oxoethylidene]amino]oxy]-](http://img.cochemist.com/ccimg/102600/102507-71-1_b.png)
![Piperidine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/97300/97231-91-9.png)
![Piperidine, 4-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/97300/97231-91-9_b.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4.png)
![Thieno[3,2-c]pyridine-5(4H)-aceticacid, a-(2-chlorophenyl)-6,7-dihydro-,methyl ester](http://img.cochemist.com/ccimg/90100/90055-48-4_b.png)
![N-[2-(dimethylamino)ethyl]acridine-4-carboxamide](http://img.cochemist.com/ccimg/89500/89459-25-6.png)
![N-[2-(dimethylamino)ethyl]acridine-4-carboxamide](http://img.cochemist.com/ccimg/89500/89459-25-6_b.png)

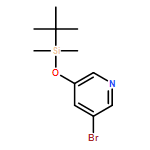
![PYRIDINE, 5-BROMO-2-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/88200/88139-92-8.png)
![PYRIDINE, 5-BROMO-2-[[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]METHYL]-](http://img.cochemist.com/ccimg/88200/88139-92-8_b.png)



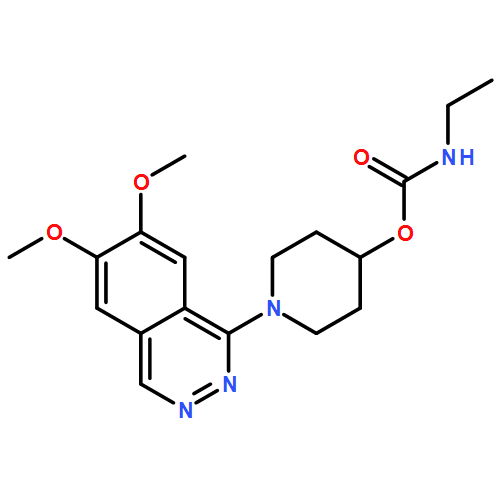
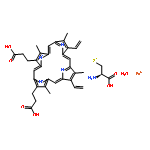
![L-gamma-glutamyl-S-[(1R)-7-(hydroxymethyl)-2,3-dihydro-1H-pyrrolizin-1-yl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/64700/64660-86-2.png)
![L-gamma-glutamyl-S-[(1R)-7-(hydroxymethyl)-2,3-dihydro-1H-pyrrolizin-1-yl]-L-cysteinylglycine](http://img.cochemist.com/ccimg/64700/64660-86-2_b.png)


![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3.png)
![5-(2-Chlorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine](http://img.cochemist.com/ccimg/55200/55142-85-3_b.png)




![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9.png)
![Octadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/51100/51063-97-9_b.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2.png)
![Hexadecanoic acid,1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/40300/40290-32-2_b.png)



![Hexadecanoic acid,1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/30400/30334-71-5.png)
![Hexadecanoic acid,1,1'-[(1S)-1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/30400/30334-71-5_b.png)


![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/17800/17738-71-5.png)
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/17800/17738-71-5_b.png)


![Octadecanoic acid, 3-hydroxy-2-[(1-oxohexadecyl)oxy]propyl ester](http://img.cochemist.com/ccimg/14100/14015-55-5.png)
![Octadecanoic acid, 3-hydroxy-2-[(1-oxohexadecyl)oxy]propyl ester](http://img.cochemist.com/ccimg/14100/14015-55-5_b.png)
![5-Chlorobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/13800/13771-75-0.png)
![5-Chlorobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/13800/13771-75-0_b.png)




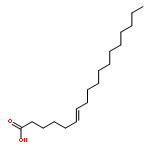
![Acetic acid,2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/4400/4332-50-7.png)
![Acetic acid,2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/4400/4332-50-7_b.png)



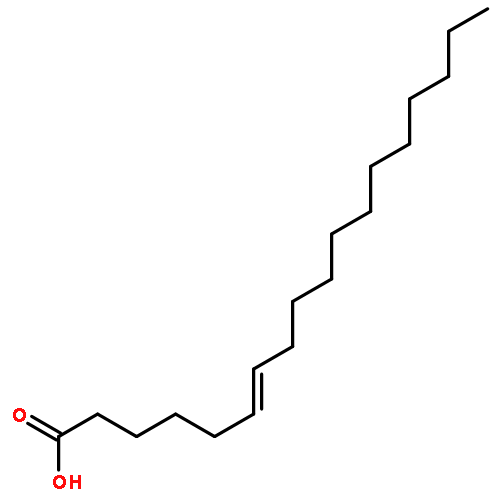
![9,12-Octadecadienoicacid (9Z,12Z)-, 1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/2500/2442-62-8.png)
![9,12-Octadecadienoicacid (9Z,12Z)-, 1,1'-[1-(hydroxymethyl)-1,2-ethanediyl] ester](http://img.cochemist.com/ccimg/2500/2442-62-8_b.png)
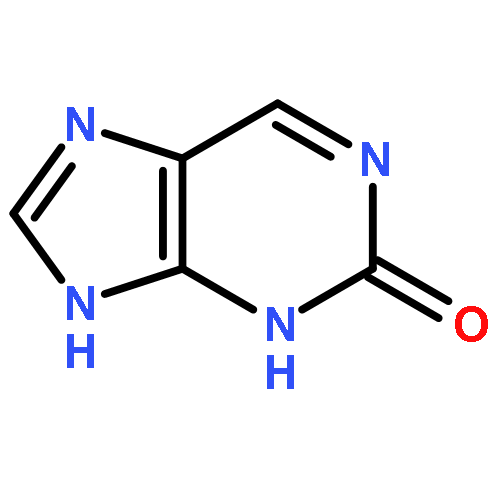

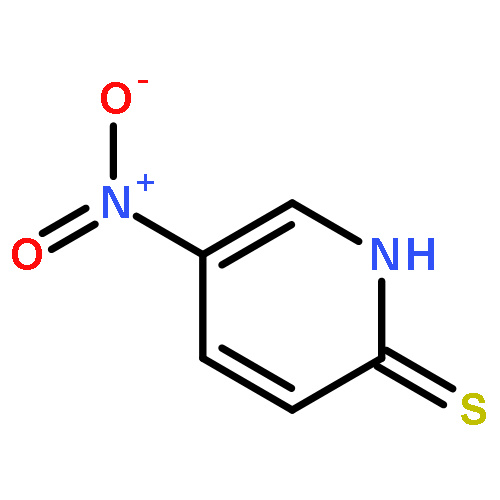




![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9.png)
![Glycine, N-[(3a,5b,7a)-3,7-dihydroxy-24-oxocholan-24-yl]-](http://img.cochemist.com/ccimg/700/640-79-9_b.png)



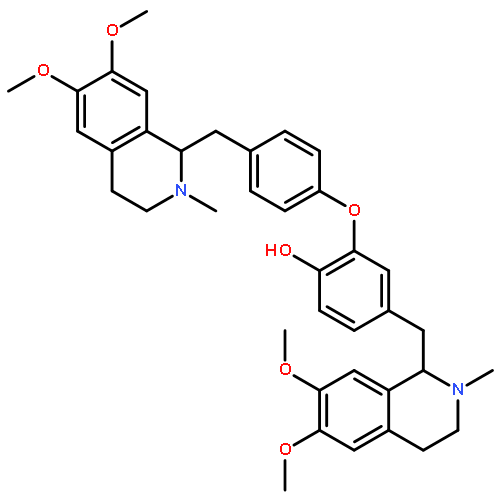
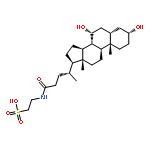

![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7.png)
![5'-O-[hydroxy(sulfooxy)phosphoryl]adenosine 3'-(dihydrogen phosphate)](http://img.cochemist.com/ccimg/500/482-67-7_b.png)


![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3Z,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/500/480-54-6.png)
![[1,6]Dioxacyclododecino[2,3,4-gh]pyrrolizine-2,7-dione,3-ethylidene-3,4,5,6,9,11,13,14,14a,14b-decahydro-6-hydroxy-6-(hydroxymethyl)-5-methyl-,(3Z,5R,6S,14aR,14bR)-](http://img.cochemist.com/ccimg/500/480-54-6_b.png)
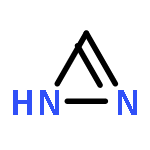
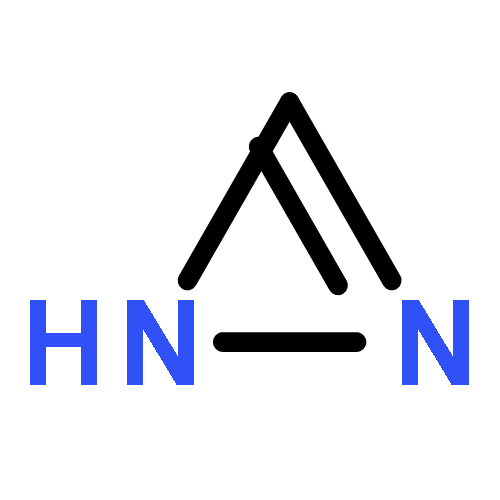






![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3.png)
![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3_b.png)