Co-reporter:Ruth Patchett, Richard C. Knighton, James D. Mattock, Alfredo Vargas, and Adrian B. Chaplin
Inorganic Chemistry November 20, 2017 Volume 56(Issue 22) pp:14345-14345
Publication Date(Web):November 2, 2017
DOI:10.1021/acs.inorgchem.7b02441
The synthesis of cationic rhodium and iridium complexes of a bis(imidazole-2-thione)-functionalized calix[4]arene ligand and their surprising capacity for potassium binding are described. In both cases, uptake of the alkali metal into the calix[4]arene cavity occurs despite adverse electrostatic interactions associated with close proximity to the transition-metal fragment [Rh+···K+ = 3.715(1) Å; Ir+···K+ = 3.690(1) Å]. The formation and constituent bonding of these unusual heterobimetallic adducts have been interrogated through extensive solution and solid-state characterization, examination of the host–guest chemistry of the ligand and its upper-rim unfunctionalized calix[4]arene analogue, and use of density functional theory based energy decomposition analysis.
Co-reporter:Simone A. Hauser;Jack Emerson-King;Scott Habershon
Chemical Communications 2017 vol. 53(Issue 26) pp:3634-3636
Publication Date(Web):2017/03/28
DOI:10.1039/C6CC09807J
Iridium(I) carbonyl complex [Ir(2,6-(PtBu2CH2)2C6H3)(CO)] undergoes reversible C–H bond activation of benzene and a series of fluorobenzenes on UV irradiation. Exclusive ortho-selectivity is observed in reactions of fluorobenzene and 1,2-difluorobenzene.
Co-reporter:Sebastian D. Pike;Mark R. Crimmin
Chemical Communications 2017 vol. 53(Issue 26) pp:3615-3633
Publication Date(Web):2017/03/28
DOI:10.1039/C6CC09575E
Fluorobenzenes, in particular fluorobenzene (FB) and 1,2-difluorobenzene (1,2-DiFB), are increasingly becoming recognised as versatile solvents for conducting organometallic chemistry and transition-metal-based catalysis. The presence of fluorine substituents reduces the ability to donate π-electron density from the arene and consequently fluorobenzenes generally bind weakly to metal centres, allowing them to be used as essentially non-coordinating solvents or as readily displaced ligands. In this context, examples of well-defined complexes of fluorobenzenes are discussed, including trends in binding strength with increasing fluorination and different substitution patterns. Compared to more highly fluorinated benzenes, FB and 1,2-DiFB typically demonstrate greater chemical inertness, however, C–H and C–F bond activation reactions can be induced using appropriately reactive transition metal complexes. Such reactions are surveyed, including catalytic examples, not only to provide perspective for the use of FB and 1,2-DiFB as innocent solvent media, but also to highlight opportunities for their exploitation in contemporary organic synthesis.
Co-reporter:Richard C. Knighton, Adrian B. Chaplin
Tetrahedron 2017 Volume 73, Issue 31(Issue 31) pp:
Publication Date(Web):3 August 2017
DOI:10.1016/j.tet.2017.06.023
The synthesis of four new dissymmetric cavitands is reported. These deep-walled receptors are constructed from a resorcin[4]arene scaffold bearing anti-disposed quinoxaline substituents, with either N-haloalkyl-diazaphthalimide (1), dinitrophenyl (2) or diaminophenyl (3) moieties as the other wall components. The structure and inclusion properties of 1 and 2 have been probed in solution by NMR spectroscopy and notably in the solid-state by X-ray crystallography. The diazaphthalimide-based compounds 1 crystallise as 1:1 host-guest complexes with chloroform, with the resorcin[4]arene scaffolds adopting pinched cone conformations. Conversely, the dinitrophenyl-variant 2 features a more open, symmetric structure in the solid-state and co-crystallises with two acetone molecules within the central cavity. Preliminary binding experiments in mesitylene-d12 at 303 K demonstrate 1 (Kapp = 5 × 102 M−1) and 2 (Kapp = 2 × 102 M−1) are effective hosts for cyclohexane guest molecules in the absence of competitive solvent inclusion.Download high-res image (205KB)Download full-size image
Co-reporter:Lucero González-Sebastián, Adrian B. Chaplin
Inorganica Chimica Acta 2017 Volume 460(Volume 460) pp:
Publication Date(Web):24 April 2017
DOI:10.1016/j.ica.2016.08.006
•A series of imidazolinium-based CCC pro-ligands have been prepared.•The corresponding free carbenes have been characterised in situ by NMR spectroscopy.•Three dinuclear copper adducts, [(CCC)(CuCl)2], have been isolated.•Two iridium pincer complexes, [Ir(CCC)HCl(NCMe)], have been isolated.A series of imidazolinium-based CCC pro-ligands featuring N-Mes, Dipp, iPr and tBu substituents (1·2HCl) have been prepared. The corresponding free carbenes are readily generated through deprotonation by strong bases and, in addition to being characterised in situ by 1H and 13C NMR spectroscopy, were trapped through reaction with CuCl. Iridium pincer compounds of the N-Mes (5a) and N-Dipp (5b) substituted ligands, viz. [Ir(1)HCl(NCMe)], were obtained through reaction between the respective pro-ligand, [Ir(COE)2Cl]2, and Et3N in acetonitrile at ca. 80 °C. Under similar conditions the N-iPr and N-tBu analogues were not formed. The new iridium pincer complexes 5a and 5b were fully characterised in solution, by NMR spectroscopy and ESI-MS, and in the solid-state by X-ray diffraction. Under relatively forcing reaction conditions neither 5a nor 5b, in combination with KOtBu, show any significant catalytic activity for the transfer dehydrogenation of cyclooctane to cyclooctene using tert-butylethylene as the sacrificial hydrogen acceptor (ca. 2 TONs, 150 °C, 24 h).Download high-res image (72KB)Download full-size image
Co-reporter:Jack Emerson-King;Richard C. Knighton;Matthew R. Gyton
Dalton Transactions 2017 vol. 46(Issue 35) pp:11645-11655
Publication Date(Web):2017/09/11
DOI:10.1039/C7DT02648J
In the context of advancing the use of metal-based building blocks for the construction of mechanically interlocked molecules, we herein describe the preparation of late transition metal containing [2]rotaxanes (1). Capture and subsequent retention of the interlocked assemblies are achieved by the formation of robust and bulky complexes of rhodium(III) and iridium(III) through hydrogenation of readily accessible rhodium(I) and iridium(I) complexes [M(COD)(PPh3)2][BArF4] (M = Rh, 2a; Ir, 2b) and reaction with a bipyridyl terminated [2]pseudorotaxane (3·db24c8). This work was underpinned by detailed mechanistic studies examining the hydrogenation of 1 : 1 mixtures of 2 and bipy in CH2Cl2, which proceeds with disparate rates to afford [M(bipy)H2(PPh3)2][BArF4] (M = Rh, 4a[BArF4], t = 18 h @ 50 °C; Ir, 4b[BArF4], t < 5 min @ RT) in CH2Cl2 (1 atm H2). These rates are reconciled by (a) the inherently slower reaction of 2a with H2 compared to that of the third row congener 2b, and (b) the competing and irreversible reaction of 2a with bipy, leading to a very slow hydrogenation pathway, involving rate-limiting substitution of COD by PPh3. On the basis of this information, operationally convenient and mild conditions (CH2Cl2, RT, 1 atm H2, t ≤ 2 h) were developed for the preparation of 1, involving in the case of rhodium-based 1a pre-hydrogenation of 2a to form [Rh(PPh3)2]2[BArF4]2 (8) before reaction with 3·db24c8. In addition to comprehensive spectroscopic characterisation of 1, the structure of iridium-based 1b was elucidated in the solid-state using X-ray diffraction.
Co-reporter:Jack Emerson-King;Simone A. Hauser
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 4) pp:787-789
Publication Date(Web):2017/01/25
DOI:10.1039/C6OB02556K
The N-heterocyclic carbene IBioxMe4 enacts selective single and double C–F bond activation of octafluorotoluene and hexafluorobenzene, respectively. The formation of the fluoroarene substituted, zwitterionic imidazoliumolate products is consistent with a mechanism involving nucleophilic aromatic substitution and subsequent oxazoline ring opening by liberated fluoride.
Co-reporter:Rhiann E. Andrew, Caroline M. Storey and Adrian B. Chaplin
Dalton Transactions 2016 vol. 45(Issue 21) pp:8937-8944
Publication Date(Web):09 May 2016
DOI:10.1039/C6DT01263A
With a view to use as carbene transfer agents, well-defined silver(I) and copper(I) complexes of a macrocyclic NHC-based pincer ligand, bearing a central lutidine donor and a dodecamethylene spacer [CNC–(CH2)12, 1], have been prepared. Although the silver adduct is characterised by X-ray diffraction as a dinuclear species anti-[Ag(μ-1)]22+, variable temperature measurements indicate dynamic structural interchange in solution involving fragmentation into mononuclear [Ag(1)]+ on the NMR time scale. In contrast, a mononuclear structure is evident in both solution and the solid-state for the analogous copper adduct partnered with the weakly coordinating [BArF4]− counter anion. A related copper derivative, bearing instead the more coordinating cuprous bromide dianion [Cu2Br4]2−, is notable for the adoption of an interesting tetranuclear assembly in the solid-state, featuring two cuprophilic interactions and two bridging NHC donors, but is not retained on dissolution. Coinage metal precursors [M(1)]n[BArF4]n (M = Ag, n = 2; M = Cu, n = 1) both act as carbene transfer agents to afford palladium, rhodium and nickel complexes of 1 and the effectiveness of these precursors has been evaluated under equivalent reaction conditions.
Co-reporter:Rhiann E. Andrew, Lucero González-Sebastián and Adrian B. Chaplin
Dalton Transactions 2016 vol. 45(Issue 4) pp:1299-1305
Publication Date(Web):17 Dec 2015
DOI:10.1039/C5DT04429D
In this frontier article we overview the emergence and scope of NHC-based CCC and CNC pincer systems, i.e. complexes containing mer-tridentate ligands bearing two NHC donor groups, comment on their effectiveness in applications, and highlight areas for future development and exploitation.
Co-reporter:Ruth Patchett and Adrian B. Chaplin
Dalton Transactions 2016 vol. 45(Issue 21) pp:8945-8955
Publication Date(Web):10 May 2016
DOI:10.1039/C6DT01001F
The preparation and coordination chemistry of 5,17-bis(3-methyl-1-imidazol-2-ylidene)-25,26,27,28-tetrapropoxycalix[4]arene (1) is described. Starting from the bis(imidazolium) pro-ligand 1·2HI, the free carbene 1 was readily generated in solution through deprotonation using K[OtBu] and its reactivity with rhodium(I) dimers [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) and [Rh(CO)2Cl]2 investigated. Dinuclear complexes were isolated in both cases, where the calix[4]arene-based NHC ligand adopts a bridging μ2-coordination mode, and in one case characterised in the solid-state by X-ray diffraction. Using instead an isolated and well-defined (mononuclear) silver transfer agent, generated by reaction of 1·2HI with Ag2O in the presence of a halide extractor, reactions with [Rh(COD)Cl]2 and [Rh(CO)2Cl]2 produced cationic dinuclear complexes bearing μ2-1 and μ2-Cl bridging ligands. The structural formulation of the novel dinuclear adducts of 1 was aided through spectroscopic congruence with model complexes, containing monodentate 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (IiPr2Me2).
Co-reporter:Dr. Thibault Troadec;Sze-yin Tan;Dr. Christopher J. Wedge;Dr. Jonathan P. Rourke;Dr. Patrick R. Unwin;Dr. Adrian B. Chaplin
Angewandte Chemie International Edition 2016 Volume 55( Issue 11) pp:3754-3757
Publication Date(Web):
DOI:10.1002/anie.201511467
Abstract
Oxidation of zero-valent phosphine complexes [M(PtBu3)2] (M=Pd, Pt) has been investigated in 1,2-difluorobenzene solution using cyclic voltammetry and subsequently using the ferrocenium cation as a chemical redox agent. In the case of palladium, a mononuclear paramagnetic PdI derivative was readily isolated from solution and fully characterized (EPR, X-ray crystallography). While in situ electrochemical measurements are consistent with initial one-electron oxidation, the heavier congener undergoes C−H bond cyclometalation and ultimately affords the 14 valence-electron PtII complex [Pt(κ2PC-PtBu2CMe2CH2)(PtBu3)]+ with concomitant formation of [Pt(PtBu3)2H]+.
Co-reporter:Dr. Thibault Troadec;Sze-yin Tan;Dr. Christopher J. Wedge;Dr. Jonathan P. Rourke;Dr. Patrick R. Unwin;Dr. Adrian B. Chaplin
Angewandte Chemie 2016 Volume 128( Issue 11) pp:3818-3821
Publication Date(Web):
DOI:10.1002/ange.201511467
Abstract
Oxidation of zero-valent phosphine complexes [M(PtBu3)2] (M=Pd, Pt) has been investigated in 1,2-difluorobenzene solution using cyclic voltammetry and subsequently using the ferrocenium cation as a chemical redox agent. In the case of palladium, a mononuclear paramagnetic PdI derivative was readily isolated from solution and fully characterized (EPR, X-ray crystallography). While in situ electrochemical measurements are consistent with initial one-electron oxidation, the heavier congener undergoes C−H bond cyclometalation and ultimately affords the 14 valence-electron PtII complex [Pt(κ2PC-PtBu2CMe2CH2)(PtBu3)]+ with concomitant formation of [Pt(PtBu3)2H]+.
Co-reporter:Simone A. Hauser, Ivan Prokes and Adrian B. Chaplin
Chemical Communications 2015 vol. 51(Issue 21) pp:4425-4428
Publication Date(Web):13 Feb 2015
DOI:10.1039/C5CC00529A
Interaction of the reactive 14 VE {Ir(IBioxMe4)3}+ fragment with fluoroarenes results exclusively in ortho-C–H bond oxidative addition and formation of 16 VE Ir(III) derivatives [Ir(IBioxMe4)3(Ar)H]+ (Ar = 2-C6H4F, 2,3-C6H3F2, 2,4,6-C6H2F3). The C–H bond activation reactions occur under mild conditions and are reversible.
Co-reporter:Rhiann E. Andrew
Inorganic Chemistry 2015 Volume 54(Issue 1) pp:312-322
Publication Date(Web):December 10, 2014
DOI:10.1021/ic5024828
Using a general synthetic procedure employing readily accessed terminal alkene-functionalized pro-ligands and macrocyclization by ring-closing olefin metathesis, rhodium carbonyl complexes have been prepared that contain lutidine (1a; n = 1) and pyridine (1b; n = 0) derived tridentate CNC macrocycles with dodecamethylene spacers. In solution, 1a shows temperature-invariant time-averaged C2 symmetry by 1H NMR spectroscopy (CD2Cl2, 500 MHz), whereas in the solid-state, two polymorphs can be obtained showing different conformations of the alkyl spacer about the metal–carbonyl bond (asymmetric and symmetric). In contrast, time-averaged motion of alkyl spacer in 1b can be halted by cooling below 225 K (CD2Cl2, 500 MHz), and the complex crystallizes as a dimer with an interesting unsupported Rh···Rh bonding interaction (3.2758(6) Å). Oxidative addition reactions of 1a and 1b, using MeI and PhICl2, have been studied in situ by 1H NMR spectroscopy, although pure Rh(III) adducts can be ultimately isolated only with the pyridine-based macrocyclic ligand. The lutidine backbone of 1a can be deprotonated by addition of K[N(SiMe3)2], and the resulting neutral dearomatized complex (5) has been fully characterized in solution, by variable-temperature 1H NMR spectroscopy, and in the solid state, by X-ray diffraction.
Co-reporter:Thibault Troadec, Amber L. Thompson, Adrian B. Chaplin
Tetrahedron Letters 2015 Volume 56(Issue 31) pp:4613-4615
Publication Date(Web):29 July 2015
DOI:10.1016/j.tetlet.2015.05.110
An operationally simple and high yielding sodium-template-based synthesis of a large bis(pyridine)-based macrocycle is described. Complexation of two equivalents of 2,6-bis(10-undecen-1-oxymethyl)pyridine with Na[BArF4] (ArF = 3,5-C6H3(CF3)2) enabled the selective cross-metathesis of the terminal alkene substituents and formation of the corresponding bis(pyridine) macrocycle in 51% isolated yield. The new 54-membered macrocycle was characterized in the solid-state by X-ray diffraction in addition to verification of the structure in solution by NMR spectroscopy and ESI-MS.
Co-reporter:Rhiann E. Andrew, Dominic W. Ferdani, C. André Ohlin, and Adrian B. Chaplin
Organometallics 2015 Volume 34(Issue 5) pp:913-917
Publication Date(Web):February 18, 2015
DOI:10.1021/om501292k
Reversible interaction with carbon monoxide results in the onset of dynamic atropisomerism at 298 K in an otherwise static NHC-based rhodium pincer complex, [Rh(C^N^C-(CH2)12)(CO)][BArF4] (1, ArF = 3,5-C6H3(CF3)2). The mechanism of this process has been comprehensively interrogated by a combination of variable-temperature NMR spectroscopy, IR spectroscopy, and computational modeling. In addition, a structural analogue of a high-energy symmetrical intermediate species—invoked in the process but not directly observed spectroscopically—has been prepared and characterized in solution and the solid-state.
Co-reporter:Simone A. Hauser, Ralf Tonner, and Adrian B. Chaplin
Organometallics 2015 Volume 34(Issue 17) pp:4419-4427
Publication Date(Web):September 2, 2015
DOI:10.1021/acs.organomet.5b00658
A method for accessing the formally 14 VE iridium(III) hydride fragment {Ir(IBioxMe4)2(H)2}+ (2), containing the conformationally rigid NHC ligand IBioxMe4, is reported. Hydrogenation of trans-[Ir(IBioxMe4)2(COE)Cl] (1) in the presence of excess Na[BArF4] leads to the formation of dimeric [{Ir(IBioxMe4)2(H)2}2Cl][BArF4] (3), which is structurally fluxional in solution and acts as a reservoir of monomeric 2 in the presence of excess halogen ion abstractor. Stable dihydride complexes trans-[Ir(IBioxMe4)2(2,2′-bipyridine)(H)2][BArF4] (4) and [Ir(IBioxMe4)3(H)2][BArF4] (5) were subsequently isolated through in situ trapping of 2 using 2,2′-bipyridine and IBioxMe4, respectively, and fully characterized. Using mixtures of 3 and Na[BArF4] as a latent source of 2, the reactive monomeric fragment’s reactivity was explored with excess ethylene and cyclooctene, and trans-[Ir(IBioxMe4)2(C2H4)2][BArF4] (6) and cis-[Ir(IBioxMe4)2(COD)][BArF4] (7) were isolated, respectively, through sacrificial hydrogenation of the alkenes. Complex 6 is notable for the adoption of a very unusual orthogonal arrangement of the trans-ethylene ligands in the solid state, which has been analyzed computationally using energy and charge decomposition (EDA-NOCV). The formation of 7 via transfer dehydrogenation of COE highlights the ability to partner IBioxMe4 with reactive metal centers capable of C–H bond activation, without intramolecular activation. Reaction of 7 with CO slowly formed trans-[Ir(IBioxMe4)2(CO)2][BArF4] (8), but the equivalent reaction with bis-ethylene 6 was an order of magnitude faster, quantifying the strong coordination of COD in 7.
Co-reporter:Jan-Niclas Luy, Simone A. Hauser, Adrian B. Chaplin, and Ralf Tonner
Organometallics 2015 Volume 34(Issue 20) pp:5099-5112
Publication Date(Web):September 23, 2015
DOI:10.1021/acs.organomet.5b00692
Computational methods have been used to analyze distorted coordination geometries in a coherent range of known and new rhodium(I) and iridium(I) complexes containing bioxazoline-based NHC ligands (IBiox). Such distortions are readily placed in context of the literature through measurement of the Cnt(NHC)–CNCN–M angle (ΘNHC; Cnt = ring centroid). On the basis of restricted potential energy calculations using cis-[M(IBioxMe4)(CO)2Cl] (M1; M = Rh, Ir), in-plane (yawing) tilting of the NHC was found to incur significantly steeper energetic penalties than orthogonal out-of-plane (pitching) movement, which is characterized by noticeably flat potential energy surfaces. Energy decomposition analysis (EDA) of the ground-state and pitched structures of M1 indicated only minor differences in bonding characteristics. In contrast, yawing of the NHC ligand is associated with a significant increase in Pauli repulsion (i.e., sterics) and reduction in M→NHC π back donation, but is counteracted by supplemental stabilizing bonding interactions only possible due to the closer proximity of the methyl substituents with the metal and ancillary ligands. Aided by this analysis, comparison with a range of carefully selected model systems and EDA, distorted coordination modes in trans-[M(IBioxMe4)2(COE)Cl] (M2; M = Rh, Ir) and [M(IBioxMe4)3]+ (M3; M = Rh, Ir) have been rationalized. Steric interactions were identified as the major contributing factor and are associated with a high degree of NHC pitching. In the case of Rh3, weak agostic interactions also contribute to the distortions, particularly with respect to NHC yawing, and are notable for increasing the bond dissociation energy of the distorted ligands. Supplementing the computational analysis, an analogue of the formally 14 VE Rh(I) species Rh3 bearing the cyclohexyl-functionalized IBiox6 ligand ([Rh(IBiox6)3]+, Rh3-Cy) was prepared and found to exhibit an exceptionally distorted NHC ligand (ΘNHC = 155.7(2)°) in the solid state.
Co-reporter:James E. Wheatley, C. André Ohlin and Adrian B. Chaplin
Chemical Communications 2014 vol. 50(Issue 6) pp:685-687
Publication Date(Web):21 Nov 2013
DOI:10.1039/C3CC48015A
Reaction of [Ir(COD)(py–ItBu)]+ (py–ItBu = 3-tert-butyl-1-picolylimidazol-2-ylidene) with acetonitrile results in reversible intramolecular C–H bond activation of the NHC ligand and formation of [Ir(η2:η1-C8H13)(py–ItBu′)(NCMe)]+. Coordinated COD acts as an internal hydride acceptor and acetonitrile coordination offsets the otherwise unfavourable thermodynamics of the process.
Co-reporter:Adrian B. Chaplin
Organometallics 2014 Volume 33(Issue 3) pp:624-626
Publication Date(Web):January 23, 2014
DOI:10.1021/om401223w
The isolation, characterization and reactivity of a T-shaped rhodium(I) complex containing Glorius’ bioxazoline derived N-heterocyclic carbene ligand IBioxMe4 is described: [Rh(IBioxMe4)3][BArF4] (1). 1 represents a rare example of a solution-stable “naked” 14-electron complex and is characterized in the solid state by highly distorted ligand geometries and Rh···C distances >3.1 Å for the IBioxMe4 alkyl substituents. Consistent with the bulky nature of the NHC ligand, no reaction was observed with excess IBioxMe4, PCy3, or norbornadiene. Reaction of 1 with CO, however, led to coordinatively saturated [Rh(IBioxMe4)3(CO)][BArF4] (2).
Co-reporter:Adrian B. Chaplin
Organometallics 2014 Volume 33(Issue 12) pp:3069-3077
Publication Date(Web):June 2, 2014
DOI:10.1021/om500332n
The preparation and characterization of a series of mono-, bis-, and tris-ligated rhodium(I) complexes of Glorius’ conformationally rigid bioxazoline-derived N-heterocyclic carbene ligand IBioxMe4 are described. Through reaction of [Rh(COE)2Cl]2 (COE = cis-cyclooctene) with isolated IBioxMe4, [Rh(IBioxMe4)(COE)Cl]2 (1), trans-[Rh(IBioxMe4)2(COE)Cl] (2), and [Rh(IBioxMe4)3Cl] (3) were each isolated by careful choice of the reaction conditions. Further substitution and salt metathesis reactions of 1–3 were investigated, and derivatives containing CO, norbornadiene, and cyclopentadienyl ancillary ligands were readily isolated. Notably, halide abstraction from 2 and 3 using Na[BArF4] (ArF = 3,5-C6H3(CF3)2) resulted in the formation of low-coordinate T-shaped cis-[Rh(IBioxMe4)2(COE)][BArF4] (9) and [Rh(IBioxMe4)3][BArF4] (11). The solid-state structures of 2, 9, and 11 each feature IBioxMe4 ligands that bind unusually with tilted coordination geometries.
Co-reporter:Simone A. Hauser, Ivan Prokes and Adrian B. Chaplin
Chemical Communications 2015 - vol. 51(Issue 21) pp:NaN4428-4428
Publication Date(Web):2015/02/13
DOI:10.1039/C5CC00529A
Interaction of the reactive 14 VE {Ir(IBioxMe4)3}+ fragment with fluoroarenes results exclusively in ortho-C–H bond oxidative addition and formation of 16 VE Ir(III) derivatives [Ir(IBioxMe4)3(Ar)H]+ (Ar = 2-C6H4F, 2,3-C6H3F2, 2,4,6-C6H2F3). The C–H bond activation reactions occur under mild conditions and are reversible.
Co-reporter:Simone A. Hauser, Jack Emerson-King, Scott Habershon and Adrian B. Chaplin
Chemical Communications 2017 - vol. 53(Issue 26) pp:NaN3636-3636
Publication Date(Web):2017/03/17
DOI:10.1039/C6CC09807J
Iridium(I) carbonyl complex [Ir(2,6-(PtBu2CH2)2C6H3)(CO)] undergoes reversible C–H bond activation of benzene and a series of fluorobenzenes on UV irradiation. Exclusive ortho-selectivity is observed in reactions of fluorobenzene and 1,2-difluorobenzene.
Co-reporter:Sebastian D. Pike, Mark R. Crimmin and Adrian B. Chaplin
Chemical Communications 2017 - vol. 53(Issue 26) pp:NaN3633-3633
Publication Date(Web):2017/03/17
DOI:10.1039/C6CC09575E
Fluorobenzenes, in particular fluorobenzene (FB) and 1,2-difluorobenzene (1,2-DiFB), are increasingly becoming recognised as versatile solvents for conducting organometallic chemistry and transition-metal-based catalysis. The presence of fluorine substituents reduces the ability to donate π-electron density from the arene and consequently fluorobenzenes generally bind weakly to metal centres, allowing them to be used as essentially non-coordinating solvents or as readily displaced ligands. In this context, examples of well-defined complexes of fluorobenzenes are discussed, including trends in binding strength with increasing fluorination and different substitution patterns. Compared to more highly fluorinated benzenes, FB and 1,2-DiFB typically demonstrate greater chemical inertness, however, C–H and C–F bond activation reactions can be induced using appropriately reactive transition metal complexes. Such reactions are surveyed, including catalytic examples, not only to provide perspective for the use of FB and 1,2-DiFB as innocent solvent media, but also to highlight opportunities for their exploitation in contemporary organic synthesis.
Co-reporter:Rhiann E. Andrew, Lucero González-Sebastián and Adrian B. Chaplin
Dalton Transactions 2016 - vol. 45(Issue 4) pp:NaN1305-1305
Publication Date(Web):2015/12/17
DOI:10.1039/C5DT04429D
In this frontier article we overview the emergence and scope of NHC-based CCC and CNC pincer systems, i.e. complexes containing mer-tridentate ligands bearing two NHC donor groups, comment on their effectiveness in applications, and highlight areas for future development and exploitation.
Co-reporter:Rhiann E. Andrew and Adrian B. Chaplin
Dalton Transactions 2014 - vol. 43(Issue 3) pp:NaN1423-1423
Publication Date(Web):2013/10/31
DOI:10.1039/C3DT52578C
A series of macrocyclic CNC pincer pro-ligands based on bis(imidazolium)lutidine salts with octa-, deca- and dodecamethylene spacers have been prepared and their coordination chemistry investigated. Using a Ag2O based transmetallation strategy, cationic palladium(II) chloride complexes [PdCl{CNC–(CH2)n}][BArF4] (n = 8, 10, 12; ArF = 3,5-C6H3(CF3)2) were prepared and fully characterised in solution, by NMR spectroscopy and ESI-MS, and in the solid-state, by X-ray crystallography. The smaller macrocyclic complexes (n = 8 and 10) exhibit dynamic behaviour in solution, involving ring flipping of the alkyl spacer across the Pd–Cl bond, which was interrogated by variable temperature NMR spectroscopy. In the solid-state, distorted coordination geometries are observed with the spacer skewed to one side of the Pd–Cl bond. In contrast, a static C2 symmetric structure is observed for the dodecamethylene based macrocycle. For comparison, palladium(II) fluoride analogues [PdF{CNC–(CH2)n}][BArF4] (n = 8, 10, 12) were also prepared and their solution and solid-state structures contrasted with those of the chlorides. Notably, these complexes exhibit very low frequency 19F chemical shifts (ca. −400 ppm) and the presence of C–H⋯F interactions (2hJFC coupling observed by 13C NMR spectroscopy). The dynamic behaviour of the fluoride complexes is largely consistent with the smaller ancillary ligand; [PdF{CNC–(CH2)8}][BArF4] exceptionally shows C2v time averaged symmetry in solution at room temperature (CD2Cl2, 500 MHz) as a consequence of dual fluxional processes of the pincer backbone and alkyl spacer.
Co-reporter:Jack Emerson-King, Simone A. Hauser and Adrian B. Chaplin
Organic & Biomolecular Chemistry 2017 - vol. 15(Issue 4) pp:NaN789-789
Publication Date(Web):2016/12/22
DOI:10.1039/C6OB02556K
The N-heterocyclic carbene IBioxMe4 enacts selective single and double C–F bond activation of octafluorotoluene and hexafluorobenzene, respectively. The formation of the fluoroarene substituted, zwitterionic imidazoliumolate products is consistent with a mechanism involving nucleophilic aromatic substitution and subsequent oxazoline ring opening by liberated fluoride.
Co-reporter:Rhiann E. Andrew, Caroline M. Storey and Adrian B. Chaplin
Dalton Transactions 2016 - vol. 45(Issue 21) pp:NaN8944-8944
Publication Date(Web):2016/05/09
DOI:10.1039/C6DT01263A
With a view to use as carbene transfer agents, well-defined silver(I) and copper(I) complexes of a macrocyclic NHC-based pincer ligand, bearing a central lutidine donor and a dodecamethylene spacer [CNC–(CH2)12, 1], have been prepared. Although the silver adduct is characterised by X-ray diffraction as a dinuclear species anti-[Ag(μ-1)]22+, variable temperature measurements indicate dynamic structural interchange in solution involving fragmentation into mononuclear [Ag(1)]+ on the NMR time scale. In contrast, a mononuclear structure is evident in both solution and the solid-state for the analogous copper adduct partnered with the weakly coordinating [BArF4]− counter anion. A related copper derivative, bearing instead the more coordinating cuprous bromide dianion [Cu2Br4]2−, is notable for the adoption of an interesting tetranuclear assembly in the solid-state, featuring two cuprophilic interactions and two bridging NHC donors, but is not retained on dissolution. Coinage metal precursors [M(1)]n[BArF4]n (M = Ag, n = 2; M = Cu, n = 1) both act as carbene transfer agents to afford palladium, rhodium and nickel complexes of 1 and the effectiveness of these precursors has been evaluated under equivalent reaction conditions.
Co-reporter:James E. Wheatley, C. André Ohlin and Adrian B. Chaplin
Chemical Communications 2014 - vol. 50(Issue 6) pp:NaN687-687
Publication Date(Web):2013/11/21
DOI:10.1039/C3CC48015A
Reaction of [Ir(COD)(py–ItBu)]+ (py–ItBu = 3-tert-butyl-1-picolylimidazol-2-ylidene) with acetonitrile results in reversible intramolecular C–H bond activation of the NHC ligand and formation of [Ir(η2:η1-C8H13)(py–ItBu′)(NCMe)]+. Coordinated COD acts as an internal hydride acceptor and acetonitrile coordination offsets the otherwise unfavourable thermodynamics of the process.
Co-reporter:Ruth Patchett and Adrian B. Chaplin
Dalton Transactions 2016 - vol. 45(Issue 21) pp:NaN8955-8955
Publication Date(Web):2016/05/10
DOI:10.1039/C6DT01001F
The preparation and coordination chemistry of 5,17-bis(3-methyl-1-imidazol-2-ylidene)-25,26,27,28-tetrapropoxycalix[4]arene (1) is described. Starting from the bis(imidazolium) pro-ligand 1·2HI, the free carbene 1 was readily generated in solution through deprotonation using K[OtBu] and its reactivity with rhodium(I) dimers [Rh(COD)Cl]2 (COD = 1,5-cyclooctadiene) and [Rh(CO)2Cl]2 investigated. Dinuclear complexes were isolated in both cases, where the calix[4]arene-based NHC ligand adopts a bridging μ2-coordination mode, and in one case characterised in the solid-state by X-ray diffraction. Using instead an isolated and well-defined (mononuclear) silver transfer agent, generated by reaction of 1·2HI with Ag2O in the presence of a halide extractor, reactions with [Rh(COD)Cl]2 and [Rh(CO)2Cl]2 produced cationic dinuclear complexes bearing μ2-1 and μ2-Cl bridging ligands. The structural formulation of the novel dinuclear adducts of 1 was aided through spectroscopic congruence with model complexes, containing monodentate 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene (IiPr2Me2).





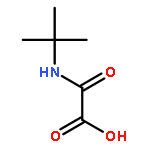
![Acetic acid, [[2,6-bis(1-methylethyl)phenyl]amino]oxo-](http://img.cochemist.com/ccimg/169800/169772-26-3.png)
![Acetic acid, [[2,6-bis(1-methylethyl)phenyl]amino]oxo-](http://img.cochemist.com/ccimg/169800/169772-26-3_b.png)


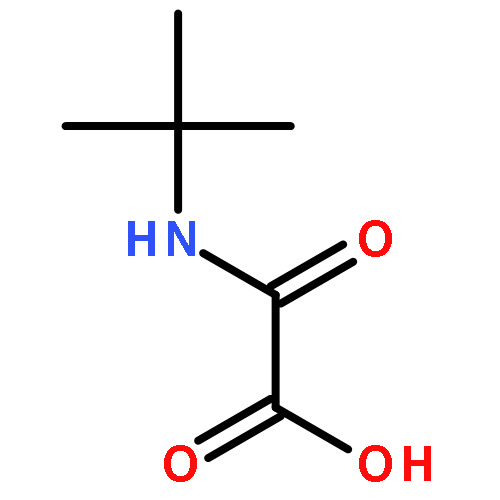
![ACETIC ACID, OXO[(2,4,6-TRIMETHYLPHENYL)AMINO]-](http://img.cochemist.com/ccimg/79400/79354-46-4.png)
![ACETIC ACID, OXO[(2,4,6-TRIMETHYLPHENYL)AMINO]-](http://img.cochemist.com/ccimg/79400/79354-46-4_b.png)


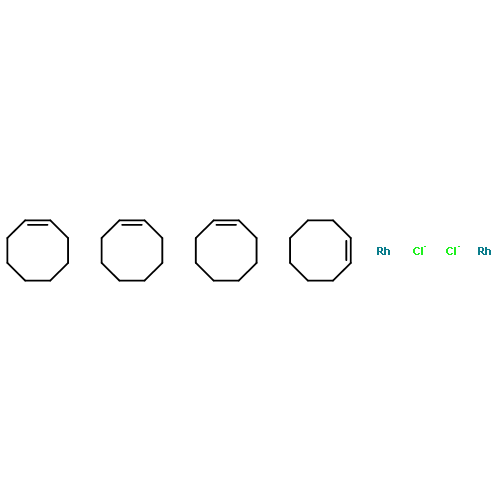
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0.png)
![bis[(2,3,5,6-η)-bicyclo[2.2.1]hepta-2,5-diene]di-μ-chlorodirhodium](http://img.cochemist.com/ccimg/12300/12257-42-0_b.png)