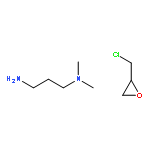
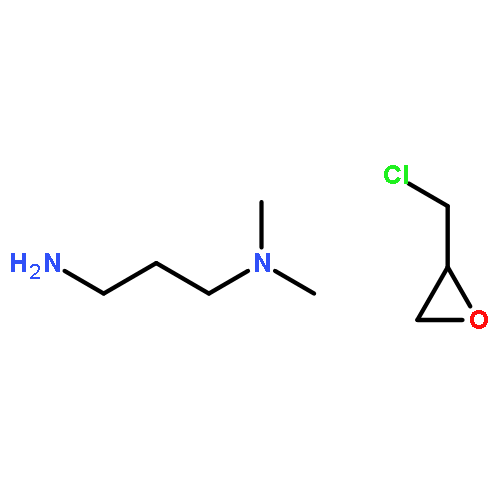
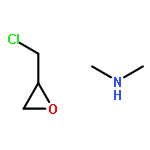
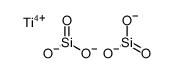Co-reporter:Yueqiang Cao, Zhijun Sui, Yian Zhu, Xinggui Zhou, and De Chen
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7835-7835
Publication Date(Web):October 10, 2017
DOI:10.1021/acscatal.7b01745
Highly dispersed bimetallic Pd-In catalysts on Al2O3 were prepared by a simple impregnation method. In comparison with the unsupported intermetallic catalyst, the supported Pd-In catalyst exhibited several magnitudes higher activity and similar selectivity for selective acetylene hydrogenation. Moreover, the activity, selectivity, and anticoking performance of the Pd-In catalyst were superior to those of the monometallic Pd catalyst. The electron transferred from indium weakened the adsorption of ethylene on the negatively charged Pd sites and hence improved the selectivity of Pd-In/Al2O3. The inhibited formation of hydride due to the presence of indium also contributed to the higher selectivity. The promoted activation of hydrogen, owing to the weak adsorption of acetylene on Pd-In/Al2O3, and decreased particle size jointly contributed to the enhanced activity of Pd-In/Al2O3. In addition, green oil formation on Pd-In/Al2O3 was retarded by the presence of indium, contributing to the enhanced stability of the catalyst. The bimetallic Pd-In catalysts showed a strongly composition dependent performance, which resulted from the different extent of electronic and/or geometric modification of Pd active sites.Keywords: acetylene hydrogenation; green oil; hydride; indium; Pd/Al2O3;
Co-reporter:Ping Li;Qian Zhao;De Chen;Weikang Yuan
The Journal of Physical Chemistry C January 29, 2009 Volume 113(Issue 4) pp:1301-1307
Publication Date(Web):Publication Date (Web): January 8, 2009
DOI:10.1021/jp808474c
Carbon nanofibers (CNFs) were immobilized on carbon microfiber (CMF) felt by chemical vapor deposition with Ni catalysts. A carbon coating, which was derived from a viscoid phenolic resin thin film involved with Ni salts, was introduced as a protect layer to stabilize the Ni nanoparticles for CNF growth, and as an interface to enhance the interaction between the CNFs and the CMFs, thus improving the uniformity and anchorage of the CNFs on the felt. The uniformly dispersed Ni nanoparticles in the carbon coating homogenously covering the CMF surfaces have resulted in a uniform layer of grown CNFs throughout the felt. The interlocked network of the entangled CNFs combined with possible penetration of the roots of CNFs into the carbon coating has enhanced the anchorage of CNFs on the surface of CMFs.
Co-reporter:Xiang Feng, Nan Sheng, Yibin Liu, Xiaobo Chen, De Chen, Chaohe Yang, and Xinggui Zhou
ACS Catalysis April 7, 2017 Volume 7(Issue 4) pp:2668-2668
Publication Date(Web):February 27, 2017
DOI:10.1021/acscatal.6b03498
It is a challenging task to design efficient Au/Ti-containing catalysts for propene epoxidation with H2 and O2 that simultaneously achieve high catalytic stability and selectivity to propylene oxide (PO). This contribution first describes the synthesis of a nanocrystalline mesoporous titanium silicalite-1 (MTS-1) by dry-gel conversion that employs cheap triblock copolymers as template. Compared with TS-1, MTS-1 has a shortened reactant/product diffusion length as a result of smaller crystal size (<100 nm) and the presence of mesopores (ca. 3 nm). Surprisingly, this designed catalyst shows simultaneously improved PO selectivity over 95% as well as stability over 40 h, much better than the traditional Au/TS-1 catalyst. Moreover, the intrinsic reason for the enhanced performance is elucidated. The better mass transfer ability together with higher hydrophobicity of Au/MTS-1 results in lower coke weight and absence of refractory aromatic coke. In this way, the side reactions and deactivation caused by blocking of micropore are inhibited.Keywords: Au/TS-1; mass transfer; propene epoxidation; selectivity; stability;
Co-reporter:Jingwei Zheng;Jiajia Ding;Dongliang Jin;Guanghua Ye;Kake Zhu;Weimin Yang;Weikang Yuan
Chemical Communications 2017 vol. 53(Issue 45) pp:6132-6135
Publication Date(Web):2017/06/01
DOI:10.1039/C7CC03403B
Phenyltrimethoxysilane as a Si source can significantly slow down the crystallization process for SAPO-34 synthesis, leading to the formation of agglomerated nanocrystals (<100 nm). The obtained nanosized SAPO-34 shows enhanced catalytic stability in methanol-to-olefin conversion under industrially relevant conditions.
Co-reporter:Guanghua Ye, Yuanyuan Sun, Xinggui Zhou, Kake Zhu, Jinghong Zhou, Marc-Olivier Coppens
Chemical Engineering Journal 2017 Volume 329(Volume 329) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.cej.2017.02.036
•An adaptable, efficient pore network cutting algorithm is proposed.•Pore networks with 8 archetypical shapes are successfully built as examples.•Pore networks are applied to simulate diffusion and reaction in porous catalysts.•Shape and randomness of the pore network could affect catalytic performance.•A larger effectiveness factor of catalysts is found for a regular pore network.A method is established to generate pore networks within domains of arbitrary shape, as long as the domain can be mathematically described by a set of inequalities. In this method, a stochastic network algorithm is adopted to construct pore network skeletons, which are then cut into the desired shapes using a new pore network cutting algorithm. The latter can be embedded into other methods to transplant its ‘pore network cutting’ function. Using this method, pore networks with four archetypical two-dimensional shapes (namely, cross-sections of one-holed rings, trilobes, four-holed rings, and wheels) and four three-dimensional shapes (namely, spheres, cylinders, trilobes, and hollow cylinders) are constructed as examples. Then, some of these pore networks are applied to simulate diffusion and reaction in Pd/γ-alumina catalyst particles for hydrogenation of benzene to cyclohexane. It is shown that the randomness of the pore network and the external particle shape significantly affect the performance of catalysts, because of their impact on effective diffusivity and diffusion length, respectively, indicating that this structural information must be accounted for to achieve a model with high accuracy. The versatile method proposed in this article is ideal to study the effect of particle shape and pore network structure on the performance of porous materials for catalysis and other applications.Download high-res image (167KB)Download full-size image
Co-reporter:X. Feng, D. Chen and X. G. Zhou
RSC Advances 2016 vol. 6(Issue 50) pp:44050-44056
Publication Date(Web):26 Apr 2016
DOI:10.1039/C6RA05772A
A tetrapropyl-ammonium (TPA) template, as the most critical component inside microporous channels of an uncalcined Au/TS-1 catalyst (Au/TS-1-B), hinders the entrance of Au nanoparticles into the micropores and thus prevents the micropore blocking induced deactivation for direct propene epoxidation with H2 and O2. In this work, the essential stability of the TPA template at distinct reaction temperatures is first investigated. It is found that the TPA template inside micropores decomposes to H2O, NH3 and propene after 350 °C. Based on a suitable reaction temperature range (<350 °C), the PO formation rate shows a volcano-shaped relationship with reaction temperature and reaches a maximum value of 220 gPO h−1 kgCat−1 at 260 °C, which is a noteworthy result compared with other reported catalysts. Moreover, due to presence of the TPA template inside micropores of TS-1, size-dependent selectivity shown on Au/TS-1-B should be intrinsic because the Au/TS-1-B catalyst exhibits uniform-sized Au nanoparticles and high stability. Hence, a series of Au/TS-1-B catalysts with different Au sizes are then tested for propene epoxidation, and the plausible reaction routes are also discussed.
Co-reporter:Zhiting Liu, Xuezhi Duan, Hongye Cheng, Jinghong Zhou, Xinggui Zhou, Weikang Yuan
Carbon 2015 Volume 89() pp:93-101
Publication Date(Web):August 2015
DOI:10.1016/j.carbon.2015.03.022
Graphite oxide (GO) with varied oxidation degrees and hexachloroplatinic acid were used as precursors for the synthesis of Pt/graphene composites via a polyol approach. It is revealed that the epoxy-rich basal planes of GO serve as the active sites for the nucleation of Pt nanoparticles via COPt bonds. With the increased oxidation degree of GO, both the nucleation sites and the repulsive forces between the GO and hexachloroplatinic anions and between the graphene and the negatively charged Pt nanoparticles increase, and this kind of subtle balance leads to a first decrease and then an increase of Pt size and size distribution on the graphene as well as the decrease of Pt loading. Moreover, adjusting the pH in the aging stage significantly increases the Pt loading both on the basal planes and at the edges of graphene, which is attributed to the introduction of positively charged sites by π sites on the basal planes of graphene and the protonation of carbonyl and carboxyl groups at the edges.
Co-reporter:Yicheng Zhang, Zhongyan Deng, Kake Zhu and Xinggui Zhou
CrystEngComm 2015 vol. 17(Issue 17) pp:3214-3218
Publication Date(Web):25 Mar 2015
DOI:10.1039/C4CE02559H
A dry gel derived from a conventional hydrogel (or an ultrasonically treated hydrogel) containing a low amount of tetraethylammonium hydroxide (TEAOH) was subjected to a vapor-phase transport procedure using morpholine and produced SAPO-34 around 300–500 nm (or below 100 nm). The formation of small crystals has mainly benefited from the increase in tetra-coordinated Al atoms in the dry gel promoted by heating and/or ultrasonic treatment.
Co-reporter:Zhongyan Deng, Yicheng Zhang, Jingwei Zheng, Kake Zhu and Xinggui Zhou
New Journal of Chemistry 2015 vol. 39(Issue 10) pp:7777-7780
Publication Date(Web):17 Aug 2015
DOI:10.1039/C5NJ01410G
Glycerol-mediated crystallization transforms a steam-treated dry gel used as the mesoporous precursor into a hierarchical ZSM-5 zeolite. The hierarchical ZSM-5 zeolite with auxiliary mesoporosity and acidity exhibits higher activity in the aldol condensation reaction of benzaldehyde with n-butyl alcohol.
Co-reporter:Pengkai Xu, Xuezhi Duan, Gang Qian, Xing-Gui Zhou
Powder Technology 2015 284() pp: 326-335
Publication Date(Web):November 2015
DOI:10.1016/j.powtec.2015.06.054
Co-reporter:Yicheng Zhang, Kake Zhu, Xuezhi Duan, Xinggui Zhou and Weikang Yuan
Journal of Materials Chemistry A 2014 vol. 2(Issue 43) pp:18666-18676
Publication Date(Web):21 Aug 2014
DOI:10.1039/C4TA02325K
The epichlorohydrin-N,N-dimethyl-1,3-diaminopropane copolymer (PCA) has been introduced for the first time as a meso-template to successfully synthesize a hierarchical ZSM-5 zeolite (PCA-ZSM-5) with mesopores of 7–50 nm in diameter by using small-sized nanoblocks. However, when its structural analogue epichlorohydrin–dimethylamine polyamine (PCS) is used, the obtained ZSM-5 zeolite (PCS-ZSM-5) has lower mesoporosity than ZSM-5 nanocrystallite aggregates (NCA-ZSM-5) synthesized without the meso-template. The templating effect of the two employed cationic polymers (PCS and PCA) on the synthesis of hierarchical ZSM-5 is valuable and interesting to be evaluated, because they are easily available and have common structural characteristics that their macromolecular structure will be largely endangered by the decomposition of quaternary ammonium groups in the long polymer chain. PCA entrapped in the zeolite partially retains its cationic charges and macromolecular structure under hydrothermal conditions and thus has a significant templating effect on the synthesis of hierarchical ZSM-5, which is benefited from the cationic centers widely separated by more than 3 carbons in PCA. However, PCS decomposes severely into small amine molecules, due to the short separation of cationic centers. Further investigation into the templating effect of PCA shows that the small-sized and negatively charged nanoblocks can easily wrap and assemble with PCA and transform into hierarchical ZSM-5 templated by PCA. However, when using large-sized zeolite seeds to synthesize ZSM-5, PCA shows a negligible templating effect because PCA with limited charge density and low accessibility of cationic charges has insufficient interactions with zeolite seeds. The catalytic activities of PCA-ZSM-5 and NCA-ZSM-5 for acetalization of cyclohexanone with methanol are inferior to that of PCS-ZSM-5 with the highest number of acid sites, but the catalytic activities for aldol condensation of benzaldehyde with n-butyl alcohol follow the order of PCA-ZSM-5 > NCA-ZSM-5 > PCS-ZSM-5, consistent with the order of mesoporosity.
Co-reporter:Yicheng Zhang, Kake Zhu, Xinggui Zhou and Weikang Yuan
New Journal of Chemistry 2014 vol. 38(Issue 12) pp:5808-5816
Publication Date(Web):06 Aug 2014
DOI:10.1039/C4NJ00811A
Three short-chain organosilanes, i.e., 3-aminopropyltrimethoxy-silane (APTMS), [3-(2-aminoethyl)aminopropyl]trimethoxysilane (AEAPTMS) and phenylaminopropyltrimethoxysilane (PHAPTMS), were used for the synthesis of hierarchically micro-/mesoporous ZSM-5 zeolites by steam-assisted crystallization of silanized dry gels. Corresponding to the different moieties of organosilanes, the obtained hierarchically porous ZSM-5 zeolites have different degrees of mesoporosity and exhibit spherical morphology composed of small nanounits. Among the three organosilanes, PHAPTMS with a bulky cross-section is the most excellent mesopore directing agent. Tracking the structural evolution of PHAPTMS-ZSM-5 during crystallization shows that a large number of mesopores are produced in the initial steaming, but it is confirmed that it does not occur due to the presence of PHAPTMS. This initial mesoporosity is mostly preserved by the PHAPTMS molecules which inhibit the growth of the crystalline phase and suppress the high mobility of aluminosilicate species under the steam atmosphere.
Co-reporter:Yicheng Zhang, Kake Zhu, Xuezhi Duan, Ping Li, Xinggui Zhou and Weikang Yuan
RSC Advances 2014 vol. 4(Issue 28) pp:14471-14474
Publication Date(Web):19 Mar 2014
DOI:10.1039/C3RA46646A
A hierarchical ZSM-5 zeolite with uniform mesopores has been synthesized by self-assembly of zeolite seeds capped with carboxyl-ended organosilane. The carboxylate anion enhances the inorganic–surfactant interaction and enables the formation of mesopores.
Co-reporter:Yicheng Zhang, Kake Zhu, Xinggui Zhou, Weikang Yuan
Materials Letters 2014 Volume 131() pp:214-216
Publication Date(Web):15 September 2014
DOI:10.1016/j.matlet.2014.05.197
•Good dispersion of Na+ in ethanol solution and dry gel was achieved.•Hierarchically porous zeolite Beta has been obtained by steam-assisted crystallization of HDTMS-silanized dry gel prepared in ethanol.•Long-chain HDTMS is more efficient than short-chain PHAPTMS in mesopore generation in zeolite Beta.Silanized dry gel prepared by solvent evaporation was subjected to steam treatment to synthesize hierarchically porous zeolite Beta. With well dispersed sodium cations (Na+) in ethanol solution and dry gel, zeolite Beta with high degree of crystallinity can be obtained by the steam-assisted crystallization method. Two types of organosilanes, short-chain phenylaminopropyltrimethoxysilane (PHAPTMS) and long-chain hexadecyltrimethoxysilane (HDTMS), were applied to prepare the silanized dry gel and inhibit the crystallization of zeolite Beta. PHAPTMS does not generate appreciable mesopores, whereas HDTMS can successfully create abundant mesopores ranging from 3 to 10 nm, which indicates a greater spatial separation effect of the long-chain moiety than the short-chain counterpart.
Co-reporter:Xiangyang Zhang, Zhang Zhang, Zhiqing Yang, Jian Chai, Chuan Liu, Liang Zhang, Xinggui Zhou
Fluid Phase Equilibria 2014 Volume 374() pp:20-24
Publication Date(Web):25 July 2014
DOI:10.1016/j.fluid.2014.04.008
•Solubility of lasalocid sodium in five organic solvents was first measured.•Three novel polymorphic forms of lasalocid sodium were reported.•van’t Hoff equation was used to correlate the solubility data and dissolution enthalpy and entropy.•The calculated results are in good agreement with the experimental data.The solubilities of lasalocid sodium in five organic solvents including methanol, ethanol, propan-2-ol, ethyl acetate and acetone were determined at temperatures ranging from 283.15 K to 323.15 K at atmospheric pressure using a gravimetric method and three novel polymorphic forms of lasaolocid sodium were observed. van’t Hoff equation was used to correlate the experimental solubility data. The calculated values of van’t Hoff equation were found to be in good agreement with the experimental data. Further, the dissolution enthalpy and entropy of lasalocid sodium in the corresponding solvents were calculated.
Co-reporter:Guanghua Ye;Xuezhi Duan;Zhijun Sui;Kake Zhu;Weikang Yuan
Adsorption 2014 Volume 20( Issue 7) pp:843-853
Publication Date(Web):2014 October
DOI:10.1007/s10450-014-9626-8
Average diffusivity linear driving force (AD-LDF) and concentration-dependent diffusivity linear driving force (CDD-LDF) approximations are introduced to simplify the precise model describing the concentration-dependent micropore diffusion in bidisperse sorbents, and are compared with the precise model in predicting the dynamics of a sorption process under two different perturbations (i.e., step change perturbations and sinusoidal wave perturbation) with different concentrations imposed at the exterior surface of the bidisperse sorbent. The performance of the two approximations is validated by the precise model and experiments. The AD-LDF performs better in step adsorption and CDD-LDF does better in step desorption. Under sinusoidal wave perturbation, the CDD-LDF performs better. The different levels of consistency of the two approximations with the precise model are attributed to the different definitions of the diffusivities.
Co-reporter:Xiangyu Yang, Gang Qian, Xuezhi Duan, and Xinggui Zhou
Crystal Growth & Design 2013 Volume 13(Issue 3) pp:1295-1300
Publication Date(Web):January 18, 2013
DOI:10.1021/cg301758k
The effect of l-valine on the growth of the l-alanine (011) surface from solution crystallization is studied by combining single-crystal growth experiments and molecular simulation. For the first time, an unusual promotion effect of l-valine on l-alanine crystal growth, after the initial inhibition effect, is found when the impurity concentration is higher. Through molecular simulation, it is revealed that this unusual promotion effect is due to the close interaction between l-alanine and l-valine, which repels H2O around solute molecules and therefore eliminates the negative effect of H2O on surface diffusion of l-alanine.
Co-reporter:Xuezhi Duan, Gang Qian, Yan Liu, Jian Ji, Xinggui Zhou, De Chen, Weikang Yuan
Fuel Processing Technology 2013 Volume 108() pp:112-117
Publication Date(Web):April 2013
DOI:10.1016/j.fuproc.2012.05.030
Ni catalysts are widely used to catalyze ammonia decomposition. In this study, first-principles calculations are performed to investigate the preferred adsorption sites and the adsorption energies of NHx (x = 0–3) and H, and the transition states, activation energies and rate constants of NH3 stepwise dehydrogenation and associative desorption of N on the stepped Ni(211) and the close-packed Ni(111) surfaces. The results show that the barrier of associative desorption of N, which is higher than those of NH3 stepwise dehydrogenation, on Ni(211) are 1.10 eV higher than that on Ni(111), indicating that the associative desorption of N on Ni(211) is difficult to take place. Hence the strongly adsorbed N atoms would block some of the step sites of Ni(211). The N-blocking Ni(211) surface is constructed, and then NH3 decomposition on 2N–Ni(211) is investigated. The results indicate that Ni catalysts with too many or too few step sites, which can be associated with Ni particle sizes, show low ammonia decomposition activities. Therefore, different sized Ni-based catalysts are employed to catalyze ammonia decomposition, and TOFH2 as a function of Ni particle size is correlated. Finally, the structure sensitivity of ammonia decomposition over Ni catalysts is rationally interpreted.Highlights► NH3 decomposition on Ni(111) is more active than that on Ni(211). ► Strongly adsorbed N atoms are easy to block some of Ni step sites. ► The optimum Ni particle size is around 3.1–3.6 nm for NH3 decomposition.
Co-reporter:Jie Fu, Rongchun Shen, Zhongming Shu, Xinggui Zhou, and Weikang Yuan
Industrial & Engineering Chemistry Research 2013 Volume 52(Issue 23) pp:7818-7826
Publication Date(Web):May 14, 2013
DOI:10.1021/ie400148p
A Karhunen–Loève (K–L) expansion has been utilized to numerically reconstruct the bed temperatures in different tubes of a cross-flow multitubular reactor for the partial oxidation of propylene to acrolein. The K–L expansion was performed in a two-step mode on a data set consisting of the catalyst bed temperature distribution under different operating conditions. Eigenvectors, as well as the corresponding eigenvalues and coefficients, were obtained in each step. Statistical analyses were performed on the coefficients, and the cumulative variance was used as an indication of accuracy of the reconstruction. The two-step K–L expansion, with three terms in the first step and one term in the second step, can well reconstruct the catalyst bed temperature distribution. Thus, the catalyst bed temperature distribution can be represented by three coefficients of the K–L expansion, which can be further correlated with operating variables to establish simple models for process modeling, monitoring, and control.
Co-reporter:Xiangyu Yang, Gang Qian, Xuezhi Duan, and Xinggui Zhou
Industrial & Engineering Chemistry Research 2012 Volume 51(Issue 45) pp:14845
Publication Date(Web):October 11, 2012
DOI:10.1021/ie301600n
The effects of l-valine on the lateral growth of (011) and (120) surfaces of l-alanine are studied by combining molecular simulation and experimental studies. The growth of the (120) surface is significantly inhibited by l-valine, while the growth of the (011) surface is less affected. An intrinsic impurity effectiveness parameter is proposed by taking into consideration the selectivity of impurities to adsorb on both step and terrace, which explains the different inhibition effects of an impurity on different surfaces.
Co-reporter:Xuezhi Duan, Gang Qian, Chen Fan, Yian Zhu, Xinggui Zhou, De Chen, Weikang Yuan
Surface Science 2012 Volume 606(3–4) pp:549-553
Publication Date(Web):February 2012
DOI:10.1016/j.susc.2011.11.030
First-principles calculations based on density functional theory (DFT) have been performed to study the adsorption and decomposition of NH3 on Ni(110). The adsorption sites, the adsorption energies, the transition states and the activation energies of the stepwise dehydrogenation of NH3 and the associative desorption of N are determined, and the zero point energy correction is included, which makes it possible to compute the rate constants of the elementary steps in NH3 decomposition. Combined DFT calculations and kinetic analysis at 350 K indicate that the associative desorption of N has a reaction rate lower than NHx dehydrogenation and is therefore the rate determining step. The distinctly different rate constants over Ni(110), Ni(111) and Ni(211) imply that ammonia decomposition over Ni-based catalyst is a structure-sensitive reaction.Highlights► The barrier of the associative desorption of N is higher than those of NHx (x = 1–3) dehydrogenation. ► At 350 K, the associative desorption of N is the rate determining step. ► Ni(110) is more active for NH3 decomposition than Ni(111) and Ni(211). ► NH3 decomposition over Ni-based catalysts shows a remarkable structure-sensitivity.
Co-reporter:Xiangyang Zhang, Gang Qian, and Xinggui Zhou
Journal of Chemical & Engineering Data 2012 Volume 57(Issue 11) pp:2963-2970
Publication Date(Web):September 27, 2012
DOI:10.1021/je3006453
Effects of three different organic acids including racemic malic acid, succinic acid, and citric acid, which act as an impurity on the solubility and metastable zone width of zinc lactate (ZnL2), have been studied. The results show that the presence of all examined impurities increases the solution solubility and the values of solubility increase with increasing impurity concentration. The introduction of impurity also leads to a reduction on the metastable zone width, and the reductions are pronounced when the impurity concentration increased. Further, experimental data of metastable zone width were analyzed using the expression of the Nývlt’s approach and self-consistent Nývlt-like approach, which can be expressed in the form: ln(ΔTmax/T0) = Φ + β ln b, with intercept Φ = {(1 – m)/m}ln(ΔHd/RTlim) + (1/m)ln(f/KT0) and slope β = 1/m. Here T0 and Tlim are the saturation and nucleation temperature, respectively. m is the apparent nucleation order, and K is a new nucleation constant related to the factor f defined as the number of stable nuclei per unit volume, ΔHd, the heat of dissolution and R the gas constant. Comparing to the former one, the latter approach provides a more satisfactory estimation for the metastable zone width at varying saturation temperature T0. The constant β for specific system reveals independence of the temperature, while the constant Φ increases with increasing saturation temperature. In addition, both constants are proportional to the impurity concentration. Crystal habits of final products are also influenced in the presence of impurities, but the crystal structures are barely changed.
Co-reporter:Xuezhi Duan, Jian Ji, Gang Qian, Chen Fan, Yian Zhu, Xinggui Zhou, De Chen, Weikang Yuan
Journal of Molecular Catalysis A: Chemical 2012 Volume 357() pp:81-86
Publication Date(Web):May 2012
DOI:10.1016/j.molcata.2012.01.023
Ammonia decomposition on transition metal surfaces is of great importance in energy and environmental industries. With the aim to obtain a systematic knowledge about the reaction mechanism of NH3 decomposition on the close-packed surfaces of the 3d-late transition metals, we perform first-principles calculations to determine the preferred adsorption sites and the adsorption energies of NHx (x = 0–3) and H, and identify the transition states of the NH3 stepwise dehydrogenation reactions and the N recombination reactions on the close-packed Fe(1 1 0), Co(1 1 1) and Ni(1 1 1) surfaces. The results show that the calculated adsorption energies and activated energies mainly depend on the d-band center of metal surfaces. Barrier decomposition analysis reveals that the interaction among binding species in transition states will increase the energy barrier while the bonding to the surface of the species in the initial states and the transition states will decrease the energy barrier. Moreover, a linear relationship, known as the Brϕnsted–Evans–Polanyi relation, also exists between the activation energy of the N recombination reaction and the adsorption energy of N on the close-packed surfaces of the 3d-late transition metals.Graphical abstract.Highlights► N recombination has a higher energy barrier than NHx dehydrogenation steps. ► Fe (1 1 0) surface is blocked by N* and is less active than Co (1 1 1) and Ni (1 1 1). ► A linear BEP relation also holds for Fe (1 1 0), Co (1 1 1) and Ni (1 1 1).
Co-reporter:Ming-Lei Yang, Yi-An Zhu, Chen Fan, Zhi-Jun Sui, De Chen and Xing-Gui Zhou
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 8) pp:3257-3267
Publication Date(Web):21 Jan 2011
DOI:10.1039/C0CP00341G
Self-consistent periodic slab calculations based on gradient-corrected density functional theory (DFT-GGA) have been conducted to examine the reaction network of propane dehydrogenation over close-packed Pt(111) and stepped Pt(211) surfaces. Selective C–H or C–C bond cleaving is investigated to gain a better understanding of the catalyst site requirements for propane dehydrogenation. The energy barriers for the dehydrogenation of propane to form propylene are calculated to be in the region of 0.65–0.75 eV and 0.25–0.35 eV on flat and stepped surfaces, respectively. Likewise, the activation of the side reactions such as the deep dehydrogenation and cracking of C3 derivatives depends strongly on the step density, arising from the much lower energy barriers on Pt(211). Taking the activation energy difference between propylene dehydrogenation and propylene desorption as the descriptor, we find that while step sites play a crucial role in the activation of propane dehydrogenation, the selectivity towards propylene is substantially lowered in the presence of the coordinatively unsaturated surface Pt atoms. As the sole C3 derivative which prefers the cleavage of the C–C bond to the C–H bond breaking, propyne is suggested to be the starting point for the C–C bond breaking which eventually gives rise to the formation of ethane, methane and coke. These findings provide a rational interpretation of the recent experimental observations that smaller Pt particles containing more step sites are much more active but less selective than larger particles in propane dehydrogenation.
Co-reporter:Yaojie Cao, Ping Li, Jinghong Zhou, Zhijun Sui, and Xinggui Zhou
Industrial & Engineering Chemistry Research 2011 Volume 50(Issue 15) pp:9431-9436
Publication Date(Web):June 28, 2011
DOI:10.1021/ie2004187
Fibrous carbon nanofiber composite is prepared by growing carbon nanofibers on graphite felt. Pressure drop experiments are carried out with ethanol solution to study the influence of the liquid surface tension on the permeability of the composite, and residence time distribution experiments with cyclohexane and water respectively are performed to study the effect of the fluid wettability on the flow behavior in the composite. Piston dispersion exchange (PDE) model is employed to determine the dynamic liquid holdup, the axial dispersion, and the mass transfer between the dynamic and static liquids. When the fluid is less oleophilic, less space in the CNF layer will be open for the flow, and the fluid will be more likely to slip over the carbon surface. Compared with the flow in spherical particle packing and in monolith, the mass transfer in the composite is high owing to its fibrous structure that splits the fluid into streamlets. The rate of mass transfer in water is lower than that in cyclohexane because water is only trapped in some of the large pores in the CNF layer while cyclohexane suffuses the whole layer.
Co-reporter:Qing Li;Zhijun Sui;Yian Zhu;Jinghong Zhou;De Chen
Topics in Catalysis 2011 Volume 54( Issue 13-15) pp:
Publication Date(Web):2011 September
DOI:10.1007/s11244-011-9708-8
The influences of gas compositions on the rates of coke formation over a Pt–Sn/Al2O3 catalyst are studied. The coke formed on the catalyst is characterized by thermal gravimetric analysis, IR spectroscopy, Raman spectroscopy and elemental analysis. Two kinds of coke are identified from the TPO profiles and assigned to the coke on the metal and the coke on the support, respectively. The coke formed on the metal is softer (containing more hydrogen) than that formed on the support. The rate of coke formation on the metal is weakly dependent on the propylene and hydrogen pressures but increasing with the propane pressure, while the rate of coke formation on the support is increasing with the propane and propylene pressures and decreasing with the hydrogen pressure. Based on the kinetic analysis, a mechanism for the coke formation on the Pt–Sn/Al2O3 catalyst is proposed, and the dimerization of adsorbed C3H6 is identified to be the kinetic relevant step for coke formation on the metal.
Co-reporter:Yaojie Cao, Ping Li, Jinghong Zhou, Zhijun Sui, Xinggui Zhou and Weikang Yuan
Industrial & Engineering Chemistry Research 2010 Volume 49(Issue 8) pp:3944-3951
Publication Date(Web):February 25, 2010
DOI:10.1021/ie9020446
Carbon nanofibers (CNFs) are grown on graphite fiber felt with a desired shape and dimension to form a structured carbon nanofiber composite. This CNF composite has a bimodal porous structure, containing macropores due to the intertexture of graphite fibers and mesopores due to the intertwist of CNFs. The pressure drop of the composite is derived from the convective flow of fluid through the macropores and is independent of the mesopores. Both viscous and turbulent resistance increases with the CNF’s loading. After being wetted with cyclohexane and dried in the air, the CNF’s layer shrinks and becomes smoother, and the composite has a much smaller viscous and turbulent resistance for the fluid. An extended Ergun equation is developed and is shown to be able to predict very well the pressure drop from the structural parameters that are related to the CNF’s loading, i.e., macropore porosity and expanded diameter of the graphite fibers.
Co-reporter:Feng-Liu Lou, Zhi-Jun Sui, Jun-Tong Sun, Ping Li, De Chen, Xing-Gui Zhou
Materials Letters 2010 Volume 64(Issue 6) pp:711-714
Publication Date(Web):31 March 2010
DOI:10.1016/j.matlet.2009.12.046
The use of carbon nanofibers (CNFs) as polymer additive has been impeded due to the problem associated with the dispersion of CNFs. In this study, CNFs/mica hybrids were synthesized by catalytic decomposition of CO over mica supported iron catalysts to increase their dispersity in polymer matrix. Antistatic coatings were prepared by dispersing the as-synthesized hybrids into epoxy with low shearing agitation and then casting the mixture by a coater. With the help of mica, the CNFs could be easily dispersed into the resin uniformly. The morphology of the CNFs, their distributions on mica surface and the size of mica, are discriminated to have great effect on surface resistivity of the coating.
Co-reporter:Yi-An Zhu, De Chen, Xing-Gui Zhou, Per-Olof Åstrand, Wei-Kang Yuan
Surface Science 2010 Volume 604(Issue 2) pp:186-195
Publication Date(Web):15 January 2010
DOI:10.1016/j.susc.2009.11.005
Density functional theory calculations are performed to investigate the C diffusion through the surface and subsurface of Ag/Ni(1 0 0) and reconstructed Ag/Ni(1 0 0). The calculated geometric parameters indicate the center of doped Ag is located above the Ni(1 0 0) surface owing to the size mismatch. The C binding on the alloy surface is substantially weakened, arising from the less attractive interaction between C and Ag atoms, while in the subsurface, the C adsorption is promoted as the Ag coverage is increased. The effect of substitutional Ag on the adsorption property of Ni(1 0 0) is rather short-range, which agrees well with the analysis of the projected density of states. Seven pathways are constructed to explore the C diffusion behavior on the bimetallic surface. Along the most kinetically favorable pathway, a C atom hops between two fourfold hollow sites via an adjacent octahedral site in the subsurface of reconstructed Ag/Ni(1 0 0). The “clock” reconstruction which tends to improve the surface mobility, is more favorable on the alloy surface because the c(2 × 2) symmetry is inherently broken by the Ag impurity. As a consequence, the local lattice strain induced by the C transport is effectively relieved by the Ag-enhanced surface mobility and the C diffusion barrier is lowered from 1.16 to 0.76 eV.
Co-reporter:Ming-Lei Yang, Yi-An Zhu, Chen Fan, Zhi-Jun Sui, De Chen, Xing-Gui Zhou
Journal of Molecular Catalysis A: Chemical 2010 321(1–2) pp: 42-49
Publication Date(Web):
DOI:10.1016/j.molcata.2010.01.017
Co-reporter:Xiangyu YANG, Gang QIAN, Xiangyang ZHANG, Xuezhi DUAN, Xinggui ZHOU
Chinese Journal of Chemical Engineering (February 2014) Volume 22(Issue 2) pp:221-226
Publication Date(Web):1 February 2014
DOI:10.1016/S1004-9541(14)60026-4
The crystal morphology of zinc lactate trihydrate in the absence or presence of impurities (viz. succinic acid, L-malic acid and D-malic acid) is investigated by molecular simulation based on surface docking model and COMPASS force field. Combing simulation results with our previous experimental results, it is found that the solvent mainly has an inhibition effect on the (0 0 2) surface, and succinic acid impurity will inhibit the growth of (0 0 2) and (0 1 1) surfaces while two enantiomers of malic acid impurity will inhibit the (0 0 2) and (1 0 0) surfaces, which are in good agreement with the experimental results.
Co-reporter:Zhiwei FAN, Xinggui ZHOU, Ling'ai LUO, Weikang YUAN
Chinese Journal of Chemical Engineering (February 2009) Volume 17(Issue 1) pp:175-178
Publication Date(Web):1 February 2009
DOI:10.1016/S1004-9541(09)60052-5
Seven distributors with different configurations are designed and optimized by constructal approach. Their flow distribution performance and energy dissipation are investigated and compared by computational fluid dynamics (CFD) simulation. The reliability of CFD simulation is verified by experiments on the distributor that hasall distributing rectangle channels on a plate. The results show that the symmetry of the distributing channels has decisive influence on the performance of flow distribution. Increasing the generations of channel branching will improve the flow distribution uniformity, but on the other hand increase the energy dissipation. Among all the sevenconstructal distributors, the distributor that has dichotomy configuration, Y-type junctions and straight interconnecting channels, is recommended for its better flow distribution performance and less energy dissipation.
Co-reporter:Kake Zhu, Xinggui Zhou
Current Opinion in Chemical Engineering (August 2015) Volume 9() pp:42-48
Publication Date(Web):1 August 2015
DOI:10.1016/j.coche.2015.07.009
•Pore-architecture and metal position are manipulated to facilitate mass transfer.•Pore-architecture is tailored based on orientated attachment growth.•Placing metal nanoparticles on the external surface of zeolite enhances stability.Zeolites belong to microporous solids that are widely used as adsorbents, catalysts and catalyst supports. As diffusion in the microspores is slow, it is highly desirable to develop approaches to enhance mass transfer or to avoid the negative effect from mass transfer resistance. In this perspective, we underline pore architecture control for hierarchical zeolite, and spatial locations of metal nanoparticles on zeolite. For solid acid catalysts, the construction of auxiliary porosity in crystalline zeolites leading to the formation of hierarchical zeolite is a promising solution. Most successful pore-architecture construction strategies are based on templating approaches within the classic LaMer crystallization framework, whereas our strategies are based on the non-classic orientated attachment growth mechanism in dry gel crystallization. Organic additives, such as organosilanes, can be incorporated with protozeolite particles to produce hybrid mesocrystals. Combustion of organics in the mesocrystal affords hierarchical beta zeolites, the pore-architecture of which is tunable with respect to additive selection. Such a crystallization based design of pore-architecture merits small primary crystal size and improved pore-connectivity, which have been verified to be the key factors that affect diffusion. On the other hand, for zeolite supported metal catalysts that suffer from deactivation caused by pore blocking, we propose a strategy to avoid the deactivation problem by depositing metal nanoparticles exclusively on the external surfaces of support. This can be achieved by simply leaving the structure directing agents in the pores before supporting metal particles. Side reactions caused by diffusion resistance are minimized, and moreover, the involatile byproducts, if any, do not produce additional diffusion resistance. Such a simple yet effective architecture control significantly prolongs the lifetime of Au/TS-1 catalyst in direct propene epoxidation with H2 and O2. From these demonstrations, it is important to design purpose-orientated zeolite synthesis to enhance mass transfer, for which the architecture plays a key role.
Co-reporter:Xiang Feng, Xuezhi Duan, Gang Qian, Xinggui Zhou, De Chen, Weikang Yuan
Journal of Catalysis (August 2014) Volume 317() pp:99-104
Publication Date(Web):1 August 2014
DOI:10.1016/j.jcat.2014.05.006
•Au/TS-1-B catalysts show high stability and uniform-sized Au nanoparticles.•Accurate size-dependent activity of Au catalyst is achieved using Au/TS-1-B.•PO formation rate is found to vary with average Au diameter (d) as d−2.7±0.3.•Typical Au geometry on TS-1-B is truncated cuboctahedron with top facet of (1 1 1).•Corner sites are dominant Au active sites for propene epoxidation with H2/O2.Propene epoxidation with H2/O2 is a typical structure-sensitive reaction. For a given support containing tetra-coordinated Ti species such as TS-1, the catalytic activity is greatly influenced by supported Au nanoparticle size. The identification of size-dependent activity of Au catalyst was achieved over a series of Au nanoparticles (2.6–5.1 nm) deposited on the exterior surface of TS-1 by employing uncalcined TS-1. Through this approach, we could obtain very stable Au catalysts with distinguishable and uniform-sized Au particles, which are critical for structure-sensitivity analysis. The PO formation rate (molPO s−1 molAu−1) over these catalysts was found to vary with average Au diameter (d) as d−2.7±0.3. Moreover, typical Au nanoparticles on uncalcined TS-1 appeared as truncated cuboctahedron with top facet of (1 1 1). Model calculations derived from the representative Au particle shape and the above size-dependent activity were then performed to show that corner sites of Au nanoparticles are dominant Au active sites for propene epoxidation.Graphical abstractDownload high-res image (204KB)Download full-size image
Co-reporter:Xinping ZHANG, Zhijun SUI, Xinggui ZHOU, Weikang YUAN
Chinese Journal of Chemical Engineering (August 2010) Volume 18(Issue 4) pp:618-625
Publication Date(Web):1 August 2010
DOI:10.1016/S1004-9541(10)60265-0
A heterogeneous model is developed for the regeneration of the Cr2O3/Al2O3 catalyst for the propane dehydrogenation process by considering the internal mass transfer and external mass/heat transfer during the coke combustion. Simulation shows that under practical operating conditions, multi-steady states exist for the catalyst pellets and the catalyst temperature is sensitive to gas temperature. However, at increased mass flow rate or lowered oxygen concentration, multi-steady states will not appear. Under the strong influences of film diffusion, the coke in the packed bed reactor will first be exhausted at the inlet, while if the film diffusion resistance is decreased, the position of first coke exhaustion moves toward the outlet of the reactor.
Co-reporter:Xiang-Yang Zhang, Gilles Févotte, Liang Zhong, Gang Qian, Xing-Gui Zhou, Wei-Kang Yuan
Journal of Crystal Growth (15 September 2010) Volume 312(Issue 19) pp:2747-2755
Publication Date(Web):15 September 2010
DOI:10.1016/j.jcrysgro.2010.06.005
The influence of malic acid, which acts as an impurity on the cooling crystallization of zinc lactate is investigated in this paper by monitoring the relative supersaturation and the number of crystals during crystallization. The presence of malic acid increases the solution solubility and makes the metastable zone wider; it also changes the habit of the crystal. The purity of the final products is shown to be influenced by the amount and size of seed crystals, cooling rate, seeding temperature and final temperature, but appears to depend mainly on the particle size and level of supersaturation. Residual supersaturation thresholds are observed that depend on the final temperature. A model is proposed to predict the steady-state supersaturation value from the final temperature at a given impurity concentration. This model is based on Kubota and Gibbs equations.
Co-reporter:L. Zhao, J.H. Zhou, Z.J. Sui, X.G. Zhou
Chemical Engineering Science (1 January 2010) Volume 65(Issue 1) pp:30-35
Publication Date(Web):1 January 2010
DOI:10.1016/j.ces.2009.03.026
Sorbitol hydrogenolysis was carried out over a carbon nanofiber supported ruthenium catalyst prepared by incipient wetness impregnation. The carbon nanofiber supported ruthenium catalyst was shown to have an attracting behavior when compared with a commercial activated carbon supported ruthenium catalyst, especially in terms of selectivity to glycols. The preferable hydrogen partial pressure for sorbitol hydrogenolysis was ca. 8.0 MPa, lower than that usually reported in previous works. Slightly soluble calcium hydroxide, which was used as a basic promoter, remarkably increased the selectivity to glycols, as compared with the soluble sodium hydroxide. The variation of product selectivity with catalyst amount indicated that glycerol was the initial C3 polyol product while propylene glycol was derived from glycerol. The parametric investigation was further focused on the intrinsic features of sorbitol hydrogenolysis.
Co-reporter:Wen-Xin Lu, Zhi-Jun Sui, Jing-Hong Zhou, Ping Li, De Chen, Xing-Gui Zhou
Chemical Engineering Science (1 January 2010) Volume 65(Issue 1) pp:193-200
Publication Date(Web):1 January 2010
DOI:10.1016/j.ces.2009.07.001
Carbon nanofibers (CNFs) were synthesized by CO disproportionation on iron catalyst at CO concentration between 58.3% and 75.0%, H2 concentration between 8.3% and 25.0% and reaction temperature between 833 and 913 K. The time-depending rate of CNFs growth as a function of time was determined by an on-line mass spectrometer and the morphologies of all CNFs products were observed by electronic microscopy. Not only the CNFs growth rate but also the morphology of the grown CNFs were shown to vary with the three operating variables. SEM and TEM images showed that the three-dimensional morphologies of the CNFs were twist, helical or straight and an interesting relationship between the maximal growth rate and the morphology was observed. When the growth rate was between 0.8 and 0.9 mmol/(s gcat), the CNFs were twist. As the growth rate increased to 1.0 mmol/(s gcat), more helical nanofibers appeared. Straight nanofibers were produced when the growth rate reached the level of 1.2 mmol/(s gcat). Finally when the rate of CNFs growth was high at 1.8 mmol/(s gcat), the absolute majority of the solid products was amorphous carbon coexisting with some short and thick nanofibers. Under different operating conditions, the crystal faces of the catalysts had different anisotropy properties for carbon deposition, thus producing CNFs with different morphologies.
Co-reporter:Ming-Lei Yang, Yi-An Zhu, Chen Fan, Zhi-Jun Sui, De Chen and Xing-Gui Zhou
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 8) pp:NaN3267-3267
Publication Date(Web):2011/01/21
DOI:10.1039/C0CP00341G
Self-consistent periodic slab calculations based on gradient-corrected density functional theory (DFT-GGA) have been conducted to examine the reaction network of propane dehydrogenation over close-packed Pt(111) and stepped Pt(211) surfaces. Selective C–H or C–C bond cleaving is investigated to gain a better understanding of the catalyst site requirements for propane dehydrogenation. The energy barriers for the dehydrogenation of propane to form propylene are calculated to be in the region of 0.65–0.75 eV and 0.25–0.35 eV on flat and stepped surfaces, respectively. Likewise, the activation of the side reactions such as the deep dehydrogenation and cracking of C3 derivatives depends strongly on the step density, arising from the much lower energy barriers on Pt(211). Taking the activation energy difference between propylene dehydrogenation and propylene desorption as the descriptor, we find that while step sites play a crucial role in the activation of propane dehydrogenation, the selectivity towards propylene is substantially lowered in the presence of the coordinatively unsaturated surface Pt atoms. As the sole C3 derivative which prefers the cleavage of the C–C bond to the C–H bond breaking, propyne is suggested to be the starting point for the C–C bond breaking which eventually gives rise to the formation of ethane, methane and coke. These findings provide a rational interpretation of the recent experimental observations that smaller Pt particles containing more step sites are much more active but less selective than larger particles in propane dehydrogenation.
Co-reporter:Yicheng Zhang, Kake Zhu, Xuezhi Duan, Xinggui Zhou and Weikang Yuan
Journal of Materials Chemistry A 2014 - vol. 2(Issue 43) pp:NaN18676-18676
Publication Date(Web):2014/08/21
DOI:10.1039/C4TA02325K
The epichlorohydrin-N,N-dimethyl-1,3-diaminopropane copolymer (PCA) has been introduced for the first time as a meso-template to successfully synthesize a hierarchical ZSM-5 zeolite (PCA-ZSM-5) with mesopores of 7–50 nm in diameter by using small-sized nanoblocks. However, when its structural analogue epichlorohydrin–dimethylamine polyamine (PCS) is used, the obtained ZSM-5 zeolite (PCS-ZSM-5) has lower mesoporosity than ZSM-5 nanocrystallite aggregates (NCA-ZSM-5) synthesized without the meso-template. The templating effect of the two employed cationic polymers (PCS and PCA) on the synthesis of hierarchical ZSM-5 is valuable and interesting to be evaluated, because they are easily available and have common structural characteristics that their macromolecular structure will be largely endangered by the decomposition of quaternary ammonium groups in the long polymer chain. PCA entrapped in the zeolite partially retains its cationic charges and macromolecular structure under hydrothermal conditions and thus has a significant templating effect on the synthesis of hierarchical ZSM-5, which is benefited from the cationic centers widely separated by more than 3 carbons in PCA. However, PCS decomposes severely into small amine molecules, due to the short separation of cationic centers. Further investigation into the templating effect of PCA shows that the small-sized and negatively charged nanoblocks can easily wrap and assemble with PCA and transform into hierarchical ZSM-5 templated by PCA. However, when using large-sized zeolite seeds to synthesize ZSM-5, PCA shows a negligible templating effect because PCA with limited charge density and low accessibility of cationic charges has insufficient interactions with zeolite seeds. The catalytic activities of PCA-ZSM-5 and NCA-ZSM-5 for acetalization of cyclohexanone with methanol are inferior to that of PCS-ZSM-5 with the highest number of acid sites, but the catalytic activities for aldol condensation of benzaldehyde with n-butyl alcohol follow the order of PCA-ZSM-5 > NCA-ZSM-5 > PCS-ZSM-5, consistent with the order of mesoporosity.
Co-reporter:Jingwei Zheng, Jiajia Ding, Dongliang Jin, Guanghua Ye, Kake Zhu, Xinggui Zhou, Weimin Yang and Weikang Yuan
Chemical Communications 2017 - vol. 53(Issue 45) pp:NaN6135-6135
Publication Date(Web):2017/05/12
DOI:10.1039/C7CC03403B
Phenyltrimethoxysilane as a Si source can significantly slow down the crystallization process for SAPO-34 synthesis, leading to the formation of agglomerated nanocrystals (<100 nm). The obtained nanosized SAPO-34 shows enhanced catalytic stability in methanol-to-olefin conversion under industrially relevant conditions.

![[2,3,3,3-TETRAFLUORO-2-(1,1,2,2,3,3,3-HEPTAFLUOROPROPOXY)PROPANOYL] 2,3,3,3-TETRAFLUORO-2-(1,1,2,2,3,3,3-HEPTAFLUOROPROPOXY)PROPANEPEROXOATE](http://img.cochemist.com/ccimg/56400/56347-79-6.png)
![[2,3,3,3-TETRAFLUORO-2-(1,1,2,2,3,3,3-HEPTAFLUOROPROPOXY)PROPANOYL] 2,3,3,3-TETRAFLUORO-2-(1,1,2,2,3,3,3-HEPTAFLUOROPROPOXY)PROPANEPEROXOATE](http://img.cochemist.com/ccimg/56400/56347-79-6_b.png)
![Benzoic acid,6-[(3R,4S,5S,7R)-7-[(2S,3S,5S)-5-ethyl-5-[(2R,5R,6S)-5-ethyltetrahydro-5-hydroxy-6-methyl-2H-pyran-2-yl]tetrahydro-3-methyl-2-furanyl]-4-hydroxy-3,5-dimethyl-6-oxononyl]-2-hydroxy-3-methyl-,sodium salt (1:1)](http://img.cochemist.com/ccimg/26000/25999-20-6.png)
![Benzoic acid,6-[(3R,4S,5S,7R)-7-[(2S,3S,5S)-5-ethyl-5-[(2R,5R,6S)-5-ethyltetrahydro-5-hydroxy-6-methyl-2H-pyran-2-yl]tetrahydro-3-methyl-2-furanyl]-4-hydroxy-3,5-dimethyl-6-oxononyl]-2-hydroxy-3-methyl-,sodium salt (1:1)](http://img.cochemist.com/ccimg/26000/25999-20-6_b.png)