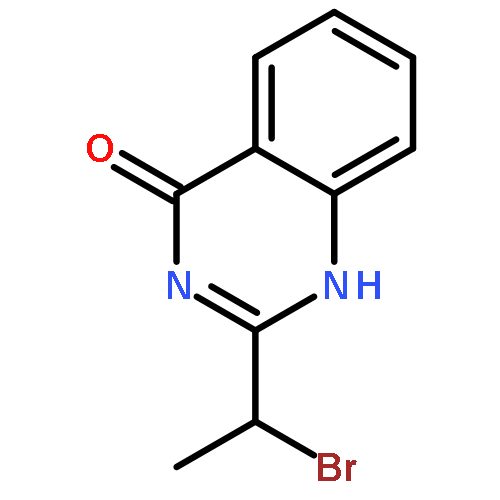
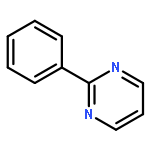
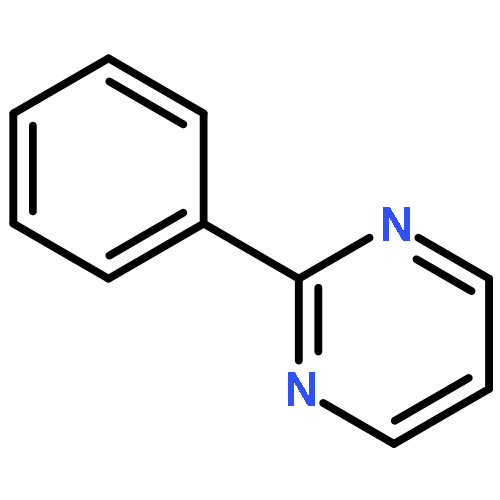
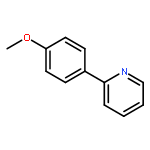
Co-reporter:Chan Shan, Jin-Wu Yan, Yu-Qing Wang, Tong Che, Zhou-Li Huang, Ai-Chun Chen, Pei-Fen Yao, Jia-Heng Tan, Ding Li, Tian-Miao OuLian-Quan Gu, Zhi-Shu Huang
Journal of Medicinal Chemistry 2017 Volume 60(Issue 4) pp:
Publication Date(Web):January 27, 2017
DOI:10.1021/acs.jmedchem.6b01218
Transcriptional control of c-myc oncogene is an important strategy for antitumor drug design. G-quadruplexes in the promoter region have been proven to be the transcriptional down-regulator of this gene. The transcriptional factor NM23-H2 can reactivate c-myc transcription by unwinding the G-quadruplex structure. Thus, down-regulation of c-myc transcription via disrupting G-quadruplex–NM23-H2 interaction might be a potential approach for cancer therapy. Here, a series of new isaindigotone derivatives were designed and synthesized based on our previous study. The abilities of these derivatives on interacting with G-quadruplexes or NM23-H2, and disrupting G-quadruplex–NM23-H2 interaction were evaluated. Among these derivatives, 19d and 22d showed remarkable abilities on disrupting G-quadruplex–NM23-H2 interaction. They exhibited significant effects on c-myc-relating processes in SiHa cells, including inhibiting the transcription and translation, inhibiting cellular proliferation, inducing apoptosis, and regulating cell cycle. Our findings provided the basis for the anticancer strategy based on c-myc transcriptional regulation via small molecules disrupting G-quadruplex–protein interaction.
Co-reporter:Chun-Li Xia, Ning Wang, Qian-Liang Guo, Zhen-Quan Liu, Jia-Qiang Wu, Shi-Liang Huang, Tian-Miao Ou, Jia-Heng Tan, Hong-Gen Wang, Ding Li, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2017 Volume 130(Volume 130) pp:
Publication Date(Web):21 April 2017
DOI:10.1016/j.ejmech.2017.02.042
•27 new 2-arylethenyl-N-methylquinolinium derivatives were synthesized as anti-AD agents.•Some of compounds showed an improved inhibitory ability on Aβ aggregation and ChEs.•Biological activities of a12 in vitro and in cellular was further evaluated.•a12 dramatically attenuated the cell death of glutamate-induced HT22 cells.A series of 2-arylethenyl-N-methylquinolinium derivatives were designed and synthesized based on our previous research of 2-arylethenylquinoline analogues as multifunctional agents for the treatment of Alzheimer's disease (AD) (Eur. J. Med. Chem. 2015, 89, 349–361). The results of in vitro biological activity evaluation, including β-amyloid (Aβ) aggregation inhibition, cholinesterase inhibition, and antioxidant activity, showed that introduction of N-methyl in quinoline ring significantly improved the anti-AD potential of compounds. The optimal compound, compound a12, dramatically attenuated the cell death of glutamate-induced HT22 cells by preventing the generation of ROS and increasing the level of GSH. Most importantly, intragastric administration of a12•HAc was well tolerated at doses up to 2000 mg/kg and could traverse blood-brain barrier.A series of compounds with an introduction of methyl group into N1-position to previous reported 2-arylethenylquinoline analogues were synthesized and evaluated. The optimal compound a12 showed good anti-AD potential.Download high-res image (235KB)Download full-size image
Co-reporter:Qian-Liang Guo, Hua-Fei Su, Ning Wang, Sheng-Rong Liao, Yu-Ting Lu, Tian-Miao Ou, Jia-Heng Tan, Ding Li, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2017 Volume 130(Volume 130) pp:
Publication Date(Web):21 April 2017
DOI:10.1016/j.ejmech.2017.02.051
•19 new acridine derivatives were synthesized as c-KIT G-quadruplexs ligands.•Some of compounds showed a good stabilizing ability on c-KIT G-quadruplexs DNA.•Introducing positive charges at 12-N position didn't improve the stabilizing ability.•Ligand 2b down-regulated the c-KIT expression and induced cell apoptosis.It has been shown that treatment of cancer cells with c-KIT G-quadruplex binding ligands can reduce their c-KIT expression levels thus inhibiting cell proliferation and inducing cell apoptosis. Herein, a series of new 7-substituted-5,6-dihydrobenzo[c]acridine derivatives were designed and synthesized. Subsequent biophysical evaluation demonstrated that the derivatives could effectively bind to and stabilize c-KIT G-quadruplex with good selectivity against duplex DNA. It was found that 12-N-methylated derivatives with a positive charge introduced at 12-position of 5,6-dihydrobenzo[c]acridine ring had similar binding affinity but lower stabilizing ability to c-KIT G-quadruplex DNA, compared with those of nonmethylated derivatives. Further molecular modeling studies showed possible binding modes of G-quadruplex with the ligands. RT-PCR assay and Western blot showed that compound 2b suppressed transcription and translation of c-KIT gene in K562 cells, which was consistent with the property of an effective G-quadruplex binding ligand targeting c-KIT oncogene promoter. Further biological evaluation showed that compound 2b could induce apoptosis through activation of the caspase-3 cascade pathway.A series of new 7-substituted-5, 6-dihydrobenzo[c]acridine derivatives were synthesized and evaluated as c-KIT G-quadruplexes DNA ligands.Download high-res image (174KB)Download full-size image
Co-reporter:Shuo-Bin Chen, Ming-Hao Hu, Guo-Cai Liu, Jin Wang, Tian-Miao Ou, Lian-Quan Gu, Zhi-Shu Huang, and Jia-Heng Tan
Journal of the American Chemical Society 2016 Volume 138(Issue 33) pp:10382-10385
Publication Date(Web):August 10, 2016
DOI:10.1021/jacs.6b04799
The RNA G-quadruplex is an important secondary structure formed by guanine-rich RNA sequences. However, its folding studies have mainly been studied in vitro. Accurate identification of RNA G-quadruplex formation within a sequence of interest remains difficult in cells. Herein, and based on the guanine-rich sequence in the 5′-UTR of NRAS mRNA, we designed and synthesized the first G-quadruplex-triggered fluorogenic hybridization (GTFH) probe, ISCH-nras1, for the unique visualization of the G-quadruplexes that form in this region. ISCH-nras1 is made up of two parts: The first is a fluorescent light-up moiety specific to G-quadruplex structures, and the second is a DNA molecule that can hybridize with a sequence that is adjacent to the guanine-rich sequence in the NRAS mRNA 5′-UTR. Further evaluation studies indicated that ISCH-nras1 could directly and precisely detect the targeted NRAS RNA G-quadruplex structures, both in vitro and in cells. Thus, this GTFH probe was a useful tool for directly investigating the folding of G-quadruplex structures within an RNA of interest and represents a new direction for the design of smart RNA G-quadruplex probes.
Co-reporter:Peng-Hui Li; Ping Zeng; Shuo-Bin Chen; Pei-Fen Yao; Yan-Wen Mai; Jia-Heng Tan; Tian-Miao Ou; Shi-Liang Huang; Ding Li; Lian-Quan Gu;Zhi-Shu Huang
Journal of Medicinal Chemistry 2016 Volume 59(Issue 1) pp:238-252
Publication Date(Web):December 9, 2015
DOI:10.1021/acs.jmedchem.5b01284
Novel topoisomerase II (Topo II) inhibitors have gained considerable interest for the development of anticancer agents. In this study, a series of 1,3-benzoazolyl-substituted pyrrolo[2,3-b]pyrazine derivatives were designed, synthesized, and evaluated as potential Topo II catalytic inhibitors. It was found that some of derivatives had good antiproliferative activity on seven cancer cell lines, especially on HL-60/MX2, a cancer cell line derivative from HL-60 that is resistant to Topo II poison. Topo II mediated DNA relaxation assay results showed that derivatives could significantly inhibit the activity of Topo II, and the structure–activity relationship studies indicated the importance of the alkylamino side chain and the benzoazolyl group. Further mechanism studies revealed that derivatives function as Topo II nonintercalative catalytic inhibitors and may block the ATP binding site of Topo II. Moreover, flow cytometric analysis showed that this class of compounds could induce apoptosis of HL-60 cells.
Co-reporter:Yin Jiang, Ai-Chun Chen, Guo-Tao Kuang, Shi-Ke Wang, Tian-Miao Ou, Jia-Heng Tan, Ding Li, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2016 Volume 122() pp:264-279
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.040
•31 new 4-anilinoquinazoline derivatives were synthesized as G-quadruplex binding ligands.•Some of compounds showed a improved interacting ability to c-myc G-quadruplex DNA.•7a was the best compound as out of the synthesized compounds.•7a significantly down-regulated c-myc gene transcription and expression in Hela cells.A series of 4-anilinoquinazoline derivatives were designed and synthesized as novel c-myc promoter G-quadruplex binding ligands. Subsequent biophysical and biochemical evaluation demonstrated that the introduction of aniline group at 4-position of quinazoline ring and two side chains with terminal amino group improved their binding affinity and stabilizing ability to G-quadruplex DNA. RT-PCR assay and Western blot showed that compound 7a could down-regulate transcription and expression of c-myc gene in Hela cells, which was consistent with the behavior of an effective G-quadruplex ligand targeting c-myc oncogene. More importantly, RTCA and colony formation assays indicated that 7a obviously inhibited Hela cells proliferation, without influence on normal primary cultured mouse mesangial cells. Flow cytometric assays suggested that 7a induced Hela cells to arrest in G0/G1 phase both in a time-dependent and dose-dependent manner.A series of compounds with an introduction of aniline group into 4-position to previous reported quinazoline derivatives were synthesized and evaluated. These compounds showed good c-myc promoter G-quadruplex binding activity and selectivity.
Co-reporter:Jia-Qiang Wu, Zhen Yang, Shang-Shi Zhang, Chun-Yong Jiang, Qingjiang Li, Zhi-Shu Huang, and Honggen Wang
ACS Catalysis 2015 Volume 5(Issue 11) pp:6453
Publication Date(Web):September 28, 2015
DOI:10.1021/acscatal.5b01801
A tandem Cp*Rh(III)-catalyzed C–H activation/Brønsted acid-catalyzed intramolecular cyclization allows a facile synthesis of carbazoles from readily available indoles. The reaction proceeds under rather mild reaction conditions with the generation of water and N2 as the only byproducts. Broad substrate scope, excellent functional group tolerance, and high yields were observed. The benzannulation of pyroles for the synthesis of indoles is also feasible using the same protocol.Keywords: Brønsted acid; carbazoles; C−H bond activation; rhodium(III); tandem catalysis
Co-reporter:Jia-Qiang Wu, Zhi-Ping Qiu, Shang-Shi Zhang, Jing-Gong Liu, Ye-Xing Lao, Lian-Quan Gu, Zhi-Shu Huang, Juan Li and Honggen Wang
Chemical Communications 2015 vol. 51(Issue 1) pp:77-80
Publication Date(Web):04 Nov 2014
DOI:10.1039/C4CC07839J
Succession of C–H activation and C–C activation was achieved by using a single rhodium(III) catalyst. Vinylcyclopropanes were used as versatile coupling partners. Mechanistic studies suggest that the olefin insertion step is rate-determining and a facile β-carbon elimination is involved, which represents a novel ring opening mode of vinylcyclopropanes.
Co-reporter:Jin-Qiang Hou, Shuo-Bin Chen, Li-Peng Zan, Tian-Miao Ou, Jia-Heng Tan, Leonard G. Luyt and Zhi-Shu Huang
Chemical Communications 2015 vol. 51(Issue 1) pp:198-201
Publication Date(Web):03 Nov 2014
DOI:10.1039/C4CC06951J
To efficiently identify small molecules binding to a G-quadruplex structure while avoiding binding to duplex DNA, we performed a multistep structure-based virtual screening by simultaneously taking into account G-quadruplex DNA and duplex DNA. Among the 13 compounds selected, one outstanding ligand shows significant selectivity for G-quadruplex binding as determined using SPR, FRET-based competition and luciferase activity assay.
Co-reporter:Jie Dai; Zhen-Quan Liu; Xiao-Qin Wang; Jing Lin; Pei-Fen Yao; Shi-Liang Huang; Tian-Miao Ou; Jia-Heng Tan; Ding Li; Lian-Quan Gu;Zhi-Shu Huang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 9) pp:3875-3891
Publication Date(Web):March 30, 2015
DOI:10.1021/acs.jmedchem.5b00139
Up-regulation of a disintegrin and metalloproteinase 10 (ADAM10) to prevent the formation of β-amyloid (Aβ) peptides might be a promising strategy to treat Alzheimer’s disease (AD). RNA G-quadruplex motif within the 5′-UTR of the ADAM10 mRNA is an inhibitory element for ADAM10 translation. Thus, mitigation of the suppressive effect of this motif using an RNA G-quadruplex-forming G-rich sequence (QGRS) binder might be a new approach for AD therapy. Herein, a series of new methylquinolinium derivatives were synthesized and screened by surface plasmon resonance (SPR) and the dual-luciferase reporter assay. Among them, compound 24 showed selective affinity for the QGRS of ADAM10 and could strongly up-regulate the translation of it. Moreover, treatment with 24 led to a significant increase of the secretion of sAPPα, consequently decreasing the Aβ40 in cellular. These results illustrate that the interaction between the RNA QGRS and a small molecule may be a new molecular strategy to modulate the translation of ADAM10.
Co-reporter:Yong Rao; Hong Liu; Lin Gao; Hong Yu; Tian-Miao Ou; Jia-Heng Tan; Shi-Liang Huang; Hong-Gen Wang; Ding Li; Lian-Quan Gu; Ji-Ming Ye;Zhi-Shu Huang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 23) pp:9395-9413
Publication Date(Web):November 17, 2015
DOI:10.1021/acs.jmedchem.5b01566
Our recent study has shown that the natural product bouchardatine (1) can reduce the triglyceride (TG) content in 3T3-L1 adipocytes (EC50 ≈ 25 μM). Here, we synthesized two series of compounds by introducing amine side chains at the 5 or 8 position of 1 and evaluated the lipid-lowering activity of derivatives. It was found that some of the compounds had significant lipid-lowering effects, and the most active compound 3d showed better activity (EC50 = 0.017 μM) than 2 (EC50 = 0.086 μM), a compound reported by us. Further, the mechanism studies revealed that 3d blocked TG accumulation via activation of the LKB1-AMPK signaling pathway, efficiently down-regulating the expression of key regulators of adipogenesis/lipogenesis. Cell uptake assay and confocal imaging of 3d in cells indicated that compound 3d had favorable cell permeability. Our results suggest that 3d may be a promising agent for the treatment of obesity and related metabolic disorders.
Co-reporter:Xiao-Qin Wang, Chun-Li Xia, Shuo-Bin Chen, Jia-Heng Tan, Tian-Miao Ou, Shi-Liang Huang, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2015 Volume 89() pp:349-361
Publication Date(Web):7 January 2015
DOI:10.1016/j.ejmech.2014.10.018
•36 new 2-arylethenylquinoline derivatives were synthesized as multifunctional anti-AD agents.•Most of compounds showed good inhibition of self-induced Aβ1–42 aggregation.•Compounds also exhibited strong antioxidation and metal chelating activity.•4b1 was the best compound as out of the synthesized compounds.A series of new 2-arylethenylquinoline derivatives (4a1–4a12, 4b1–4b8, 4c1–4c4, 4d1–4d3 and 4e1–4e9) were designed, synthesized, and evaluated as potential multifunctional agents for the treatment of Alzheimer's disease (AD). In vitro studies showed that these synthetic compounds inhibited self-induced Aβ1–42 aggregation effectively ranged from 23.6% to 83.9% at the concentration of 20 μM, and acted as potential antioxidants and biometal chelators. Their structure–activity relationships were obtained and discussed. In particular, compound 4b1, the most active compound, displayed strong inhibitory activity with an IC50 value of 9.7 μM for self-induced Aβ1–42 aggregation, good antioxidative activity with a value of 3.9-fold of Trolox, potent inhibitory activity for cholinesterase with IC50 values of 0.2 μM and 64.1 μM against butyrylcholinesterase (BuChE) and acetylcholinesterase (AChE), respectively. Besides, 4b1 was also capable of disassembling the self-induced Aβ1–42 aggregation fibrils with a ratio of 59.8% at 20 μM concentration, and had a good metal chelating activity. Taken together, these results suggest that compound 4b1 might be a promising lead compound for AD treatment.Thirty six new 2-arylethenylquinoline derivatives were synthesized as multifunctional anti-AD agents. These compounds had good activity of Aβ1–42 aggregation inhibition, cholinesterase inhibition, antioxidation, Aβ1–42 aggregation fibrils disassembly, and metal chelation. The most potential compound was 4b1. .
Co-reporter:Bing-Lei Yao, Yan-Wen Mai, Shuo-Bin Chen, Hua-Ting Xie, Pei-Fen Yao, Tian-Miao Ou, Jia-Heng Tan, Hong-Gen Wang, Ding Li, Shi-Liang Huang, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2015 Volume 92() pp:540-553
Publication Date(Web):6 March 2015
DOI:10.1016/j.ejmech.2015.01.024
•26 new benzo[a]phenazin derivatives were design and synthesized as Topo inhibitors.•Some of compounds exhibited significant Topo I and Topo II inhibitory activity.•Compounds showed excellent antiproliferative activity against HL-60 cells.•The compounds acted as Topo I poisons and Topo II catalytic inhibitors.A novel series of benzo[a]phenazin derivatives bearing alkylamino side chains were designed, synthesized and evaluated for their topoisomerases inhibitory activity as well as cytotoxicity against four human cancer cell lines (HL-60, K-562, HeLa, and A549). These compounds were found to be dual inhibitors of topoisomerase (Topo) I and Topo II, and exhibited excellent antiproliferative activity, in particular against HL-60 cells with submicromolar IC50 values. Further mechanistic studies showed that this class of compounds acted as Topo I poisons by stabilizing the Topo I-DNA cleavage complexes and Topo II catalytic inhibitors by inhibiting the ATPase activity of hTopo II. Molecular docking studies revealed the binding modes of these compounds for Topo I and Topo II.A series of benzo[a]phenazin derivatives were synthesized and evaluated for their topoisomerases inhibition and cytotoxicity. Their mechanistic studies showed that the compounds were Topo I poisons and Topo II catalytic inhibitors.
Co-reporter:Yong Rao, Hong Liu, Lin Gao, Hong Yu, Jia-Heng Tan, Tian-Miao Ou, Shi-Liang Huang, Lian-Quan Gu, Ji-Ming Ye, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 15) pp:4719-4727
Publication Date(Web):1 August 2015
DOI:10.1016/j.bmc.2015.05.057
Bouchardatine (1), a naturally occurring β-indoloquinazoline alkaloid, was synthesized. For the first time, the lipid-lowering effect and mechanism of 1 was investigated in 3T3-L1 adipocytes. Our study showed that 1 could significantly reduce lipid accumulation without cytotoxicity and mainly inhibited early differentiation of adipocyte through proliferation inhibition and cell cycle arrested in dose-dependent manner. Furthermore, the inhibition of early differentiation was reflected by down-regulation of key regulators of adipogenesis/lipogenesis, including CCAAT enhancer binding proteins (C/EBPβ, C/EBPδ, C/EBPα), peroxisome proliferator-activated receptors γ (PPARγ) and sterol-regulatory element binding protein-1c (SREBP-1c), in both of mRNA and protein levels. Subsequently decreasing the protein levels of acetyl CoA carboxylase (ACC), fatty acid synthase (FAS), and stearyl coenzyme A desaturated enzyme 1 (SCD-1), the rate-limited metabolic enzymes of fatty acid synthesis, were also observed. Further studies revealed that 1 persistently activated adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) during differentiation, suggesting that the AMPK may be an upstream mechanism for the effect of 1 on adipogenesis and lipogenesis. Our data suggest that 1 can be a candidate for the development of new therapeutic drugs against obesity and related metabolic disorders.
Co-reporter:Shuo-Bin Chen, Wei-Bin Wu, Ming-Hao Hu, Tian-Miao Ou, Lian-Quan Gu, Jia-Heng Tan and Zhi-Shu Huang
Chemical Communications 2014 vol. 50(Issue 81) pp:12173-12176
Publication Date(Web):21 Aug 2014
DOI:10.1039/C4CC05394J
A novel 2,4,5-triaryl-substituted imidazole (IZCM-1) has been found to display distinct and specific fluorescence enhancement upon binding to parallel G-quadruplexes. Such a sensitive and topology-specific probe is able to light up without affecting the topology or thermal stability of the G-quadruplex sample. Thus, these advantages distinguish IZCM-1 from other G-quadruplex probes.
Co-reporter:Jin-Wu Yan, Shuo-Bin Chen, Hui-Yun Liu, Wen-Jie Ye, Tian-Miao Ou, Jia-Heng Tan, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Chemical Communications 2014 vol. 50(Issue 52) pp:6927-6930
Publication Date(Web):07 May 2014
DOI:10.1039/C4CC01472C
A tailor-made colorimetric and red-emitting fluorescent dual probe for G-quadruplex nucleic acids was developed by incorporating a coumarin–hemicyanine fluorophore into an isaindigotone framework. The significant and distinct changes in both the color and fluorescence of this probe enable the label-free and visual detection of G-quadruplex structures.
Co-reporter:Gang Du, Su-Mei Huang, Peng Zhai, Shuo-Bin Chen, Wen-Zhao Hua, Jia-Heng Tan, Tian-Miao Ou, Shi-Liang Huang, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
Dyes and Pigments 2014 Volume 107() pp:97-105
Publication Date(Web):August 2014
DOI:10.1016/j.dyepig.2014.03.027
•New BODIPY-benzofuroquinoline conjugates were synthesized.•The conjugates can be used as fluorescence probes to determinate DNA.•The probes have high sensitivity and selectivity for double-strand DNA.The sensitive and selective detection of nucleic acids is important for basic research and many applied fields. Herein, a series of new BODIPY-benzofuroquinoline conjugates were designed, synthesized and evaluated as DNA intercalating dyes. All compounds were characterized by using 1H, 13C NMR, IR, UV–Vis and fluorescence spectroscopy, and DNA binding properties of these conjugates to calf thymus DNA were studied by using fluorescence titration, UV titration, isothermal titration calorimetry and CD analysis. Significant enhancement of the fluorescent quantum yield was observed for all the conjugates in the presence of calf thymus DNA, and one compound showed excellent sensitivity and selectivity offering its potential application as a DNA specific fluorescent probe. Our results showed that these conjugates could intercalate into calf thymus DNA with high binding affinities. The properties of these dyes as fluorescent probes for living cells imaging were also investigated.
Co-reporter:Liang Xu, Zhi-Hong Huang, Christian A. Sandoval, Lian-Quan Gu, and Zhi-Shu Huang
Organic Process Research & Development 2014 Volume 18(Issue 9) pp:1137-1141
Publication Date(Web):August 6, 2014
DOI:10.1021/op500148k
A novel efficient asymmetric hydrogenation (AH) process was developed for the preparation of (R)-1-(3,5-bis(trifluoromethyl)phenyl)ethanol (3), using a catalyst Ru/(4R,5R)-(+)-4,5-bis(diphenylphosphinomethyl)-2,2-dimethyl-1,3-dioxoane-(R,R-Diop)-2R-(α-methylmethanamine)-4,7-dimethyl-1H-benzo[d]imidazole (R-D-Me-BIMAH) in toluene in the presence of potassium t-butoxide. Various hydrogenation parameters, such as ligand, solvent, and substrate-to-catalyst (S/C) ratio, were investigated. The hydrogenation was carried out for four times on a 5 kg scale at 30 atm and 25 °C with S/C of 20 000 with an enantiomeric excess of >89%.
Co-reporter:Ying-Chun Chen, Xiao-Yi Zeng, Yan He, Hong Liu, Bin Wang, Han Zhou, Jian-Wen Chen, Pei-Qing Liu, Lian-Quan Gu, Ji-Ming Ye, and Zhi-Shu Huang
ACS Chemical Biology 2013 Volume 8(Issue 10) pp:2301
Publication Date(Web):August 20, 2013
DOI:10.1021/cb4003893
Obesity is characterized by expansion of adipose tissue, which results from an increase in adipocyte number (adipogenesis) and adipocyte size (lipogenesis). A reversal of these processes has been suggested to be a potential antiobetic therapy. Rutaecarpine (Rut) and its novel analogues (R17 and R18) were identified to exert potent effect in reducing lipid accumulation during adipocyte differentiation in 3T3-L1 adipocytes with little cytotoxicity. All three compounds reduced lipid accumulation in a dose-dependent manner, while R17 and R18 exhibited much more potent inhibitory effects compared to that of Rut. Further studies showed that R17 suppressed both adipogenesis and lipogenesis during all stages of adipocyte differentiation as indicated by the reduced protein and mRNA levels of key regulators of adipogenesis/lipogenesis, including PPARγ, C/EBPα, SREBP-1c, ACC, FAS, and SCD-1. We next examined the effect of R17 on the UPR pathway and the results showed that the UPR markers (PERK, eIF2α, IRE1α, and spliced XBP1 mRNA) were all significantly reduced by R17. Further studies revealed that R17 persistently activated AMPK during differentiation, suggesting that the AMPK may be an upstream mechanism for the effect of R17 on adipogenesis and lipogenesis via the adipogenic/lipogenic markers and the UPR pathway. Finally, studies in fast/refeeding mice demonstrated that R17 administration was able to reduce epididymal fat mass and the levels of plasma TG and FFA in vivo. Our results suggest that rutaecarpine analogues may have therapeutic potential for obesity and related metabolic disorders. The mechanism involves the suppression of adipogenic/lipogenic proteins and the suppression of the UPR pathway possibly via the AMPK.
Co-reporter:Yi-Ping Chen, Zi-Ying Zhang, Yan-Ping Li, Ding Li, Shi-Liang Huang, Lian-Quan Gu, Jun Xu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2013 Volume 66() pp:22-31
Publication Date(Web):August 2013
DOI:10.1016/j.ejmech.2013.05.015
•A series of novel isoliquiritigenin (ISL) derivatives were synthesized.•Most of synthesized compounds had better inhibition on Aβ aggregation and 5-LO.•The amide derivatives (4b–d) exhibit strong inhibitory potency against both targets.A series of new isoliquiritigenin (ISL) derivatives were synthesized and evaluated as dual inhibitors for amyloid-beta (Aβ) aggregation and 5-lipoxygenase (5-LO). It was found that all these synthetic compounds inhibited Aβ (1–42) aggregation effectively with their IC50 values ranged from 2.2 ± 1.5 μM to 23.8 ± 2.0 μM. These derivatives also showed inhibitory activity to 5-LO with their IC50 values ranged from 6.1 ± 0.1 μM to 35.9 ± 0.3 μM. Their structure–activity relationships (SAR) and mechanisms of inhibitions were studied. This study provided potentially important information for further development of ISL derivatives as multifunctional agents for Alzheimer's disease (AD) treatment.A series of new isoliquiritigenin derivatives were synthesized and evaluated as a dual inhibitor of Aβ self-induced aggregation and 5-lipoxygenase (5-LO). Compound 4d exhibited strong inhibitory potency against both targets.
Co-reporter:Shi-Tian Zhuo, Chun-Yan Li, Ming-Hao Hu, Shuo-Bin Chen, Pei-Fen Yao, Shi-Liang Huang, Tian-Miao Ou, Jia-Heng Tan, Lin-Kun An, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 24) pp:3989-4005
Publication Date(Web):04 Apr 2013
DOI:10.1039/C3OB40325D
Topoisomerases (Topo I and Topo II) are very important players in DNA replication, repair, and transcription, and are a promising class of antitumor target. In present study, a series of benzo[a]phenazine derivatives with alkylamino side chains at C-5 were designed, synthesized, and their biological activities were evaluated. Most of derivatives showed good antiproliferative activity with a range of IC50 values of 1–10 μM on the four cancer cell lines HeLa, A549, MCF-7, and HL-60. Topoisomerase-mediated DNA relaxation assay results showed that derivatives could effectively inhibit the activity of both Topo I and Topo II, and the structure–activity relationship studies indicated the importance of introducing an alkylamino side chain. Further mechanism studies revealed that the compounds could stabilize the Topo I–DNA cleavage complexes and inhibit the ATPase activity of hTopo II, indicating that they are a rare class of dual topoisomerase inhibitors by acting as Topo I poisons and Topo II catalytic inhibitors. Moreover, flow cytometric analysis and caspase-3/7 activation assay showed that this class of compounds could induce apoptosis of HL-60 cells.
Co-reporter:Zhen-Quan Liu, Shi-Tian Zhuo, Jia-Heng Tan, Tian-Miao Ou, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
Tetrahedron 2013 69(24) pp: 4922-4932
Publication Date(Web):
DOI:10.1016/j.tet.2013.04.045
Co-reporter:Chan Shan;Jia-Heng Tan;Tian-Miao Ou;Zhi-Shu Huang
Science China Chemistry 2013 Volume 56( Issue 10) pp:1351-1363
Publication Date(Web):2013 October
DOI:10.1007/s11426-013-4920-y
G-quadruplexes comprise a class of secondary structures that are formed in guanine-rich sequences in eukaryotic genomes and play a crucial role in the regulation of many biological events. G-quadruplexes have become targets for anticancer drugs with high selectivity vs. duplex DNA and low cytotoxicity against normal cells. Natural products and their derivatives display polymorphism, structural complexity, and potent activity. It is, therefore, reasonable to seek ligands targeting G-quadruplexes from natural products. Recently, many successful examples have been reported, showing ligands with excellent anticancer activities. In this review, we summarized the development of research on natural products and derivatives that target G-quadruplex structures in an effort to guide future studies.
Co-reporter:Yi Long, Zeng Li, Jia-Heng Tan, Tian-Miao Ou, Ding Li, Lian-Quan Gu, and Zhi-shu Huang
Bioconjugate Chemistry 2012 Volume 23(Issue 9) pp:1821
Publication Date(Web):August 8, 2012
DOI:10.1021/bc300123m
In order to improve the selectivity of 5-N-methyl quindoline (cryptolepine) derivatives as telomeric quadruplex binding ligands versus duplex DNA, a series of peptidyl-benzofuroquinoline (P-BFQ) conjugates (2a–2n) were designed and synthesized. Their interactions with telomeric quadruplex and duplex DNA were examined by using the fluorescence resonance energy transfer (FRET) melting assay, surface plasmon resonance (SPR), circular dichroism spectroscopy (CD), and molecular modeling studies. Introduction of a peptidyl group at 11-position of the aromatic benzofuroquinoline scaffold not only effectively increased its binding affinity, but also significantly improved its selectivity toward telomeric quadruplex versus duplex DNA. Combined with the data for their inhibitory effects on telomerase activity, their structure–activity relationships (SARs) studies showed that the types of amino acid residues and the length of the peptidyl side chains were important for the improvement of their interactions with the telomeric G-quadruplex. Long-term exposure of human cancer cells to 2c showed a remarkable cessation in population growth and cellular senescence phenotype, and accompanied by a shortening of the telomere length.
Co-reporter:Zeng Li, Jia-Heng Tan, Jin-Hui He, Yi Long, Tian-Miao Ou, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2012 Volume 47() pp:299-311
Publication Date(Web):January 2012
DOI:10.1016/j.ejmech.2011.10.057
A series of 2,4-disubstituted quinazoline derivatives found to be a new type of highly selective ligand to bind with telomeric G-quadruplex DNA, and their biological properties were reported for the first time.Their interactions with telomeric G-quadruplex DNA were evaluated by using fluorescence resonance energy transfer (FRET) melting assay, circular dichroism (CD) spectroscopy, surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), and molecular modeling. Our results showed that these derivatives could well recognize G-quadruplex and have high selectivity toward G-quadruplex over duplex DNA. The structure–activity relationships (SARs) study revealed that the disubstitution of quinazoline and the length of the amide side chain were important for its interaction with the G-quadruplex. Furthermore, telomerase inhibition of the quinazoline derivatives and their cellular effects were studied.A series of quinazoline derivatives as novel telomeric G-quadruplex ligands were synthesized and evaluated. These derivatives could well recognize G-quadruplex DNA and have significant cellular biological activity.Highlights► 17 quinazoline derivatives as novel G-quadruplex DNA ligands were synthesized. ► They showed high binding affinity and selectivity with telomeric G-quadruplex DNA. ► 11d could significantly inhibit cellular biological activity.
Co-reporter:Jin-Wu Yan, Yan-Ping Li, Wen-Jie Ye, Shuo-Bin Chen, Jin-Qiang Hou, Jia-Heng Tan, Tian-Miao Ou, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 8) pp:2527-2534
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmc.2012.02.061
A series of isaindigotone derivatives and analogues were designed, synthesized and evaluated as dual inhibitors of cholinesterases (ChEs) and self-induced β-amyloid (Aβ) aggregation. The synthetic compounds had IC50 values at micro or nano molar range for cholinesterase inhibition, and some compounds exhibited strong inhibitory activity for AChE and high selectivity for AChE over BuChE, which were much better than the isaindigotone derivatives previously reported by our group. Most of these compounds showed higher self-induced Aβ aggregation inhibitory activity than a reference compound curcumin. The structure–activity relationship studies revealed that the derivatives with higher inhibition activity on AChE also showed higher selectivity for AChE over BuChE. Compound 6c exhibiting excellent inhibition for both AChE and self-induced Aβ aggregation was further studied using CD, EM, molecular docking and kinetics.A variety of new isaindigotone derivatives were synthesized. Their structure–activity relationships were studied, and compound 6c was found to be an excellent dual inhibitors for acetylcholinesterase and amyloid beta aggregation.
Co-reporter:Wei-Jia Chen, Chen-Xi Zhou, Pei-Fen Yao, Xiao-Xiao Wang, Jia-Heng Tan, Ding Li, Tian-Miao Ou, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 9) pp:2829-2836
Publication Date(Web):1 May 2012
DOI:10.1016/j.bmc.2012.03.031
A series of 1,8-dipyrazolcarbazole (DPC) derivatives (6a–6d, 7a–7d) designed as G-quadruplex ligands have been synthesized and characterized. The FRET-melting and SPR results showed that the DPC derivatives could well recognize G-quadruplex with strong discrimination against the duplex DNA. In addition, the DPC derivatives showed much stronger stabilization activities and binding affinities for c-myc G-quadruplex rather than telomeric G-quadruplex. Therefore, their interactions with c-myc G-quadruplex were further explored by means of CD spectroscopy, PCR-stop assay, and molecular modeling. In cellular studies, all compounds showed strong cytotoxicity against cancer cells, while weak cytotoxicity towards normal cells. RT-PCR assay showed that compound 7b could down-regulate c-myc gene expression in Ramos cell line, while had no effect on c-myc expression in CA46 cell line with NHE III1 element removed, indicating its effective binding with G-quadruplex on c-myc oncogene in vivo.A series of 1,8-dipyrazolcarbazole derivatives were synthesized, and found to be a new type of selective binding ligands for c-myc G-quadruplex over duplex DNA.
Co-reporter:Jin-Qiang Hou;Shuo-Bin Chen;Jia-Heng Tan
Journal of Computer-Aided Molecular Design 2012 Volume 26( Issue 12) pp:1355-1368
Publication Date(Web):2012 December
DOI:10.1007/s10822-012-9619-1
G-quadruplexes are higher-order DNA and RNA structures formed from guanine-rich sequences. These structures have recently emerged as a new class of potential molecular targets for anticancer drugs. An understanding of the three-dimensional interactions between small molecular ligands and their G-quadruplex targets in solution is crucial for rational drug design and the effective optimization of G-quadruplex ligands. Thus far, rational ligand design has been focused mainly on the G-quartet platform. It should be noted that small molecules can also bind to loop nucleotides, as observed in crystallography studies. Hence, it would be interesting to elucidate the mechanism underlying how ligands in distinct binding modes influence the flexibility of G-quadruplex. In the present study, based on a crystal structure analysis, the models of a tetra-substituted naphthalene diimide ligand bound to a telomeric G-quadruplex with different modes were built and simulated with a molecular dynamics simulation method. Based on a series of computational analyses, the structures, dynamics, and interactions of ligand-quadruplex complexes were studied. Our results suggest that the binding of the ligand to the loop is viable in aqueous solutions but dependent on the particular arrangement of the loop. The binding of the ligand to the loop enhances the flexibility of the G-quadruplex, while the binding of the ligand simultaneously to both the quartet and the loop diminishes its flexibility. These results add to our understanding of the effect of a ligand with different binding modes on G-quadruplex flexibility. Such an understanding will aid in the rational design of more selective and effective G-quadruplex binding ligands.
Co-reporter:Tian-Miao Ou ; Jing Lin ; Yu-Jing Lu ; Jin-Qiang Hou ; Jia-Heng Tan ; Shu-Han Chen ; Zeng Li ; Yan-Ping Li ; Ding Li ; Lian-Quan Gu ;Zhi-Shu Huang
Journal of Medicinal Chemistry 2011 Volume 54(Issue 16) pp:5671-5679
Publication Date(Web):July 20, 2011
DOI:10.1021/jm200062u
G-Quadruplex is a special DNA secondary structure and present in many important regulatory regions in human genome, such as the telomeric end and the promoters of some oncogenes. Specially, different forms of G-quadruplexes exist in telomeric DNA and c-myc promoter and play important roles in the pathway of cell proliferation and senescence. The effects of G-quadruplex ligands for either telomeric or c-myc G-quadruplex in vitro have been widely studied, but the specificity of these effects in vivo is still unknown. In the present research, various experiments were carried out to study the effect of G-quadruplex ligand SYUIQ-05 on tumor cell lines and the mechanism of this effect. Our results showed that it preferred to bind with G-quadruplex in c-myc and had rather insignificant effect on G-quadruplex in telomere. Therefore, it is possible that this compound had its antitumor activity for cancer cells mainly through its interaction with c-myc quadruplex.
Co-reporter:Wei-Bin Wu, Shu-Han Chen, Jin-Qiang Hou, Jia-Heng Tan, Tian-Miao Ou, Shi-Liang Huang, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 8) pp:2975-2986
Publication Date(Web):28 Jan 2011
DOI:10.1039/C0OB00921K
A series of 2-phenyl-benzopyranopyrimidine (PBPP) derivatives with alkylamino side chains were synthesized and found to be a new type of highly selective ligand to bind with telomeric G-quadruplex DNA, and their biological properties were reported for the first time. Their interactions with telomeric G-quadruplex DNA were studied with FRET melting, surface plasmon resonance, CD spectroscopy, and molecular modeling. Our results showed that the disubstituted PBPP derivatives could strongly bind to and effectively stabilize the telomeric G-quadruplex structure, and had significant selectivity for G-quadruplex over duplex DNA. In comparison, the mono substituted derivatives had much less effect on the G-quadruplex, suggesting that the disubstitution of PBPP is essential for its interaction with the G-quadruplex. Furthermore, telomerase inhibition of the PBPP derivatives and their cellular effects were studied, and compound 11b was found to be the most promising compound as a telomerase inhibitor and telomeric G-quadruplex binding ligand for further development for cancer treatment.
Co-reporter:Wei-Bin Wu, Jie-Bin Ou, Zhi-Hong Huang, Shuo-Bin Chen, Tian-Miao Ou, Jia-Heng Tan, Ding Li, Liu-Lan Shen, Shi-Liang Huang, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 8) pp:3339-3347
Publication Date(Web):August 2011
DOI:10.1016/j.ejmech.2011.04.059
A series of mansonone F (MF) derivatives were designed and synthesized. These compounds were found to be strong inhibitors for topoisomerases, with much more significant inhibition for topoisomerase II rather than topoisomerase I. The best inhibitor showed 20 times stronger anti-topoisomerase II activity than a positive control Etoposide. The cytotoxic activity of these MF derivatives was evaluated against human cancer cell lines CNE-2 and Glc-82, which showed that these compounds were potent antitumor agents. The structure–activity relationships (SARs) study revealed that o-quinone group and pyran ring are important for their cytotoxic activity.Various novel mansonone F derivatives were designed and synthesized for SARs study, and some of these compounds showed significant stronger inhibition against topoisomerase II than a positive control Etoposide.Highlights► A series of novel mansonone F (MF) derivatives were designed and synthesized. ► The derivatives with 7,8-dione and pyran ring were particularly important for their cytotoxic activity. ► Some of these compounds showed significant inhibition against topoisomerase II, which were 20-fold more potent than a positive control Etoposide.
Co-reporter:Yan-Ping Li, Fang-Xian Ning, Meng-Bi Yang, Yong-Cheng Li, Min-Hua Nie, Tian-Miao Ou, Jia-Heng Tan, Shi-Liang Huang, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 5) pp:1572-1581
Publication Date(Web):May 2011
DOI:10.1016/j.ejmech.2011.02.005
A series of 3-substituted (5c–5f, 6c–6f) and 4-substituted (10a–10g) oxoisoaporphine derivatives were synthesized. It was found that all these synthetic compounds had IC50 values at micro or nano molar range for cholinesterase inhibition, and most of them could inhibit amyloid β (Aβ) self-induced aggregation with inhibition ratio from 31.8% to 57.6%. The structure–activity relationship studies revealed that the derivatives with higher selectivity on AChE also showed better inhibition on Aβ self-induced aggregation. The results from cell toxicity study indicated that most quaternary methiodide salts had higher IC50 values than the corresponding non-quaternary compounds. This study provided potentially important information for further development of oxoisoaporphine derivatives as lead compounds for the treatment of Alzheimer’s disease.3-substituted (5c–5f, 6c–6f) and 4-substituted (10a–10g) oxoisoaporphine derivatives were synthesized and it was found that the derivatives with higher selectivity on AChE also showed better inhibition on Aβ (1–42) self-induced aggregation.Highlights► 3-Substituted and 4-substituted oxoisoaporphine derivatives were synthesized. ► All synthetic compounds had strong inhibition on cholinesterase and Aβ aggregation. ► The derivatives with higher selectivity on AChE also showed better inhibition on Aβ self-induced aggregation.
Co-reporter:Wen Luo, Yan-Ping Li, Yan He, Shi-Liang Huang, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 6) pp:2609-2616
Publication Date(Web):June 2011
DOI:10.1016/j.ejmech.2011.03.058
A new series of heterobivalent tacrine derivatives were designed, synthesized and evaluated as potential multi-functional anti-Alzheimer agents for their inhibitory activity on cholinesterases, antioxidant activity and self-induced β-amyloid (Aβ) aggregation. All these synthesized compounds had potent inhibition activity on acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) at nanomolar range. A Lineweaver–Burk plot and molecular modeling study showed that these compounds targeted both the catalytic active site (CAS) and the peripheral anionic site (PAS) of AChE. The compounds containing hydroxyl group showed potent peroxyl radical absorbance activity. In addition, compound 5j exhibited higher self-induced Aβ aggregation inhibitory activity than curcumin, which could become a multi-functional agent for further development for the treatment of AD.A series of heterobivalent tacrine derivatives (5a–5r) were synthesized as potential multi-functional anti-Alzheimer agents with high inhibitory activity for acetylcholinesterase, butyrylcholinesterase and potent antioxidation activity. Some compounds exhibited higher self-induced Aβ aggregation inhibitory activity than curcumin.Highlights► 13 new heterobivalent tacrine derivatives as multi-functional anti-Alzheimer agents were synthesized. ► Compounds showed high inhibitory activities for AChE and BuChE. ► Compounds interacted at both catalytic active site and peripheral anionic site of AChE. ► Some of these compounds exhibited potent antioxidation activity and inhibition of self-induced Aβ aggregation.
Co-reporter:Jin-Qiang Hou, Jia-Heng Tan, Xiao-Xiao Wang, Shuo-Bin Chen, Si-Yuan Huang, Jin-Wu Yan, Shu-Han Chen, Tian-Miao Ou, Hai-Bin Luo, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 18) pp:6422-6436
Publication Date(Web):20 Jun 2011
DOI:10.1039/C1OB05884C
G-quadruplex structures are a new class of attractive targets for DNA-interactive anticancer agents. The primary building block of this structure is the G-quartet, which is composed of four coplanar guanines and serves as the major binding site for small molecules. NMR studies and molecular dynamics simulations have suggested that the planarity of G-quartet surface has been highly dynamic in solution. To better investigate how the planarity of unfused aromatic ligand impacts on its quadruplex binding properties, a variety of planarity controllable isaindigotone derivatives were designed and synthesized. The interaction of G-quadruplex DNA with these designed ligands was systematically explored using a series of biophysical studies. The FRET-melting, SPR, and CD spectroscopy results showed that reducing the planarity of their unfused aromatic core resulted in their decreased binding affinity and stabilization ability for G-quadruplex. NMR studies also suggested that these compounds could stack on the G-quartet surface. Such results are in parallel with subsequent molecular modeling studies. A detailed binding energy analysis indicated that van der Waals energy (ΔEvdw) and entropy (TΔS) are responsible for their decreased quadruplex binding and stabilization effect. All these results provided insight information about how quadruplex recognition could be controlled by adjusting the planarity of ligands, which shed light on further development of unfused aromatic molecules as optimal G-quadruplex binding ligands.
Co-reporter:Yue Li, Shu-Han Chen, Tian-Miao Ou, Jia-Heng Tan, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 6) pp:2074-2083
Publication Date(Web):15 March 2011
DOI:10.1016/j.bmc.2011.01.043
Cyclooxygenase-1/2 (COX-1/2) and 5-lipoxygenase (5-LOX) are enzymes in two different pathways in the inflammatory process. In the present study, a variety of new nimesulide derivatives were synthesized through incorporation of a 5-LOX pharmacophore into nimesulide followed with some structural modifications, which were then characterized for dual enzyme inhibitors for these two types of enzymes. Their structure–activity relationships (SARs) were studied, and compound 20f was found to be an excellent dual enzyme inhibitor. Its binding conformation and interaction mode were studied with molecular docking experiments. Compound 20f could become a lead compound for further development for potential anti-inflammatory drugs.A variety of new nimesulide derivatives were synthesized through incorporation of a 5-LOX pharmacophore into nimesulide with some further modifications. Their structure–activity relationships were studied, and compound 20f was found to be an excellent dual enzyme inhibitor.
Co-reporter:Wen Luo, Yan-Ping Li, Yan He, Shi-Liang Huang, Jia-Heng Tan, Tian-Miao Ou, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 2) pp:763-770
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmc.2010.12.022
A new series of tacrine-multialkoxybenzene hybrids (9a–9n) were designed, synthesized and evaluated as dual inhibitors of cholinesterases (ChEs) and self-induced β-amyloid (Aβ) aggregation. All the synthesized compounds had high acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) inhibitory activity with IC50 values at the nanomolar range, which were much better than tacrine alone. A Lineweaver–Burk plot and molecular modeling study showed that these hybrids targeted both the catalytic active site (CAS) and the peripheral anionic site (PAS) of AChE. Besides, compounds 9a–9f with methylenedioxybenzene moiety showed higher self-induced Aβ aggregation inhibitory activity than a reference compound, curcumin. These compounds could be selected as multi-potent agents for further investigation to treat AD.Tacrine-multialkoxybenzene hybrids were synthesized as anti-Alzheimer agents with high inhibition activity for both acetylcholinesterase, butyrylcholinesterase and amyloid beta self-aggregation.
Co-reporter:Shang-Ying Chen, Yuan Chen, Yan-Ping Li, Shu-Han Chen, Jia-Heng Tan, Tian-Miao Ou, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5596-5604
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.07.033
A series of novel curcumin analogues were designed, synthesized, and evaluated as potential multifunctional agents for the treatment of AD. The in vitro studies showed that these compounds had better inhibitory properties against Aβ aggregation than curcumin. Superior anti-oxidant properties (better than the reference compound Trolox) of these compounds were observed by the oxygen radical absorbance capacity (ORAC) method and a cell-based assay using DCFH-DA as a probe. In addition they were able to chelate metals such as iron and copper and decrease metal-induced Aβ aggregation. The structure–activity relationships were discussed. The results suggested that our curcumin analogues could be selected as multifunctional agents for further investigation of AD treatment.A series of novel curcumin analogues were designed, synthesized, and biologically evaluated as multifunctional agents targeting β-amyloid, oxidative stress, and redox-active metal ions. Compound A4 was found to have the most potent Aβ aggregation inhibitory activity (IC50 = 2.5 μM).
Co-reporter:Bin Wang, Zeng Li, Xiao Ning Wang, Jia Heng Tan, Lian Quan Gu, Zhi Shu Huang
Chinese Chemical Letters 2011 Volume 22(Issue 8) pp:951-953
Publication Date(Web):August 2011
DOI:10.1016/j.cclet.2011.01.034
A new approach to the facile synthesis of 2-substituted-quinazolin-4(3H)-ones and its derivatives using the condensation reaction of substituted 2-aminobenzamide and orthoesters is reported.
Co-reporter:Shuo-Bin Chen, Jia-Heng Tan, Tian-Miao Ou, Shi-Liang Huang, Lin-Kun An, Hai-Bin Luo, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 3) pp:1004-1009
Publication Date(Web):1 February 2011
DOI:10.1016/j.bmcl.2010.12.019
Discovery of potent and selective ligands for telomeric G-quadruplex DNA is a challenging work. Through a combination approach of pharmacophore model construction, model validation, database virtual screening, chemical synthesis and interaction evaluation, we discovered and confirmed triaryl-substituted imidazole TSIZ01 to be a new telomeric G-quadruplex ligand with potent binding and stabilizing activity to G-quadruplex DNA, as well as a 8.7-fold selectivity towards telomeric G-quadruplex DNA over duplex DNA.
Co-reporter:Yan Ma, Tian-Miao Ou, Jia-Heng Tan, Jin-Qiang Hou, Shi-Liang Huang, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2011 46(5) pp: 1906-1913
Publication Date(Web):
DOI:10.1016/j.ejmech.2011.02.020
Co-reporter:Xiao-Dong Wang ; Tian-Miao Ou ; Yu-Jing Lu ; Zeng Li ; Zheng Xu ; Chen Xi ; Jia-Heng Tan ; Shi-Liang Huang ; Lin-Kun An ; Ding Li ; Lian-Quan Gu ;Zhi-Shu Huang
Journal of Medicinal Chemistry 2010 Volume 53(Issue 11) pp:4390-4398
Publication Date(Web):May 18, 2010
DOI:10.1021/jm100445e
Human bcl-2 gene is an apoptosis-related oncogene containing a GC-rich sequence which is located upstream from P1 promoter and has the potential to form G-quadruplex structures. However, the regulatory role of the quadruplex and the effect of its ligands on bcl-2 have not been clarified. Here, we demonstrated that the G-quadruplex structure was disrupted when partial mutation of G → A was made, resulting in a 2-fold increase in basal transcriptional activity of bcl-2 promoter. Quindoline derivatives, the highly active G-quadruplex ligands developed by our group, could significantly suppress bcl-2 transcriptional activation but had less effect on mutated bcl-2 transcription. These results provided direct evidence that G-quadruplex structure formed in bcl-2 promoter region could function as a transcriptional repressor element, and G-quadruplex specific ligands could regulate the transcription of bcl-2 through stabilization of quadruplex structure. The results further indicated that quindoline derivatives could induce apoptosis of HL-60 tumor cells.
Co-reporter:Bin Wang, Yi-Chi Mai, Yue Li, Jin-Qiang Hou, Shi-Liang Huang, Tian-Miao Ou, Jia-Heng Tan, Lin-Kun An, Ding Li, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 4) pp:1415-1423
Publication Date(Web):April 2010
DOI:10.1016/j.ejmech.2009.12.044
A series of novel rutaecarpine derivatives and related alkaloid derivatives 3-aminoalkanamido-substituted rutaecarpine 4a–f and 7,8-dehydrorutaecarpine 5a–c, and 6-aminoalkanamido-substituted 3-[2-(3-Indolyl)ethyl]-4(3a)-quinazolinones 8a–c, were synthesized and subjected to pharmacological evaluation as acetylcholinesterase (AChE) inhibitors. The synthetic compounds exhibited strong inhibitory activity for AChE and high selectivity for AChE over BuChE. The structure–activity relationships were discussed and their binding conformation and simultaneous interactions mode were further clarified by kinetic characterization and the molecular docking studies.The synthesis of 5c with strong inhibitory activity for AChE and high selectivity for AChE over BuChE (ID50 = 10 nM, Selectivity Index = 539) is reported.
Co-reporter:Zeng-Yun An, Yi-Yong Yan, Dan Peng, Tian-Miao Ou, Jia-Heng Tan, Shi-Liang Huang, Lin-Kun An, Lian-Quan Gu, Zhi-Shu Huang
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 9) pp:3895-3903
Publication Date(Web):September 2010
DOI:10.1016/j.ejmech.2010.05.043
A series of graveoline and graveolinine derivatives were synthesized. The biological results showed that most of graveoline derivatives possessed higher cytotoxicity and better inhibitive effect against the adhesion and migration of human umbilical vein endothelial cell (HUVEC) than graveolinine derivatives. Among these compounds, 8d was the most potent agents that also showed significant anti-angiogenesis activities in chick embryo chorioallantoic membrane (CAM) assay.
Co-reporter:Dan Peng, Jia-Heng Tan, Shuo-Bin Chen, Tian-Miao Ou, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 23) pp:8235-8242
Publication Date(Web):1 December 2010
DOI:10.1016/j.bmc.2010.10.021
A series of bisaryldiketene derivatives were designed and synthesized as a new class of specific G-quadruplex ligands. The ligand–quadruplex interactions were further evaluated by FRET, ITC, and PCR stop assay. In contrast to most of the G-quadruplex ligands reported so far, which comprise an extended aromatic ring, these compounds are neither polycyclic nor macrocyclic, but have a non-aromatic and relative flexible linker between two quinoline moieties enabling the conformation of compounds to be flexible. Our results showed that these bisaryldiketene derivatives could selectively recognize G-quadruplex DNA rather than binding to duplex DNA. Moreover, they showed promising discrimination between different G-quadruplex DNA. The primary binding affinity of ligand M2 for c-myc G-quadruplex DNA was over 200 times larger than that for telomere G-quadruplex DNA.A series of bisaryldiketene derivatives were designed and synthesized as a new class of specific G-quadruplex ligands. The primary binding affinity of ligand M2 for c-myc G-quadruplex DNA was over 200 times larger than that for telomere G-quadruplex DNA.
Co-reporter:Jing Lin, Yi-Yong Yan, Tian-Miao Ou, Jia-Heng Tan, Shi-Liang Huang, Ding Li, Zhi-Shu Huang, and Lian-Quan Gu
Biomacromolecules 2010 Volume 11(Issue 12) pp:
Publication Date(Web):November 29, 2010
DOI:10.1021/bm100862k
G-quadruplex is a type of DNA secondary structure formed by specific guanine-rich sequences. Because of their enrichment in functional genomic regions and their biological significance, G-quadruplexes are recognized as significant drug targets for cancer and other diseases. Here, we tested the precipitation efficiency of Mg2+ for various DNA oligomers, including single-stranded, double-stranded, triplex, hairpin, i-motif, and some reported G-quadruplex DNA. It was found that Mg2+ could specifically recognize and precipitate G-quadruplex DNA with a particularly high efficiency of close to 100% for G-quadruplex structures with parallel conformation, which provided an inexpensive and convenient method for detecting and separating G-quadruplex DNA from other DNA structures as well as identifying parallel G-quadruplex from other conformational G-quadruplexes. Further experiments with both CD and gel electrophoresis validated the effectiveness of this approach. The structure of the precipitate was characterized using transmission electron microscopy (TEM), and the observed linear precipitate suggested that a polymerization of G-quadruplex DNA was formed through π−π stacking of end to end by the unique large aromatic surface of G-quadruplex.
Co-reporter:Jin-Qiang Hou, Shuo-Bin Chen, Jia-Heng Tan, Tian-Miao Ou, Hai-Bin Luo, Ding Li, Jun Xu, Lian-Quan Gu, and Zhi-Shu Huang
The Journal of Physical Chemistry B 2010 Volume 114(Issue 46) pp:15301-15310
Publication Date(Web):November 4, 2010
DOI:10.1021/jp106683n
G-quadruplexes are higher-order DNA and RNA structures formed from guanine-rich sequences, and they are attractive anticancer drug targets. Understanding the three-dimensional interactions between a G-quadruplex and its ligand in solution is the key to discovering a drug lead. Hence, from crystallographic or NMR structures, molecular dynamics studies have been performed on six ligand−quadruplex complexes. BRACO-19, BSU6039, daunomycin, RHPS4, MMQ1, and TMPyP4 are the six ligands that bind to the G-quadruplex structures in the studies. Based on molecular dynamics simulations and a series of computational analyses, the results suggest that ions move away from the external G-quartet to let the ligand bind to the quadruplex in aqueous solution. The ligand binding can increase the stability of the Hoogseen hydrogen bonds within the G-quartet. However, the G-quartet binding site can only fit one ligand molecule. The ligand can form hydrogen bonds at the loop or flank of the quadruplex. However, not all the interactions will stabilize the ligand−quadruplex complex in aqueous solution. These findings can assist in the design of selective and potent G-quadruplex ligands.
Co-reporter:Jia-Heng Tan ; Tian-Miao Ou ; Jin-Qiang Hou ; Yu-Jing Lu ; Shi-Liang Huang ; Hai-Bin Luo ; Jian-Yong Wu ; Zhi-Shu Huang ; Kwok-Yin Wong ;Lian-Quan Gu
Journal of Medicinal Chemistry 2009 Volume 52(Issue 9) pp:2825-2835
Publication Date(Web):April 8, 2009
DOI:10.1021/jm801600m
Four isaindigotone derivatives (5a,b and 6a,b) designed as telomeric G-quadruplex ligands have been synthesized and characterized. The unfused aromatic rings in these compounds allow a flexible and adaptive conformation in G-quadruplex recognition. The interaction of human telomeric G−quadruplex DNA with these designed ligands was explored by means of FRET melting, fluorescence titration, CD spectroscopy, continuous variation, and molecular modeling studies. Our results showed that the adaptive scaffold might not only allow the ligands to well occupy the G-quartet but also perfectly bind to the grooves of the G-quadruplex. The synergetic effect of the multiple binding modes might be responsible for the improved binding ability and high selectivity of these ligands toward G-quadruplex over duplex DNA. Long-term exposure of HL60 and CA46 cancer cells to compound 5a showed a remarkable decrease in population growth, cellular senescence phenotype, and shortening of the telomere length, which is consistent with the behavior of an effective telomeric G-quadruplex ligand and telomerase inhibitor.
Co-reporter:Huang Tang, Yong-Biao Wei, Chi Zhang, Fang-Xian Ning, Wei Qiao, Shi-Liang Huang, Lin Ma, Zhi-Shu Huang, Lian-Quan Gu
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 6) pp:2523-2532
Publication Date(Web):June 2009
DOI:10.1016/j.ejmech.2009.01.021
Aporphine alkaloids, isolated from Chinese medicinal herb, are important natural products. We recently reported that synthetic derivatives of oxoisoaporphine alkaloids exhibited high acetylcholinesterase inhibitory activity and high selectivity for AChE over BuChE (Bioorg. Med. Chem. Lett. 2007, 17, 3765–3768). In this paper, further research results were presented. A series of novel derivatives of oxoaporphine alkaloids (5a–j, 4-carboxylic amide-7-oxo-7H-dibenzo[de,g]quinoline, Ar-CONH(CH2)nNR) and their quaternary methiodide salts (6a–h, Ar-CONH(CH2)nN+(CH3)RI−) were designed and synthesized as acetylcholinesterase (AChE) and/or butyrylcholinesterase (BuChE) inhibitors. The AChE inhibition potency of synthetic oxoaporphine derivatives was decreased about 2–3 orders of magnitude as compared with that of oxoisoaporphine derivatives. Non-competitive binding mode was found for both kinds of derivatives. Molecular docking simulations on the oxoisoaporphine derivatives 7 series and oxoaporphine derivatives 6 series with AChE from Torpedo californica have demonstrated that the ligands bound to the dual-site of the enzyme.
Co-reporter:Yan Ma, Tian-Miao Ou, Jia-Heng Tan, Jin-Qiang Hou, Shi-Liang Huang, Lian-Quan Gu, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 13) pp:3414-3417
Publication Date(Web):1 July 2009
DOI:10.1016/j.bmcl.2009.05.030
A series of new 9-O-substituted berberine derivatives (4a–j) as telomeric quadruplex ligands was synthesized and evaluated. The results from biophysical and biochemical assay indicated that introducing of positive charged aza-aromatic terminal group into the side chain of 9-position of berberine significantly improved the binding ability with G-quadruplex, and exhibited the inhibitory effect on the hybridization and on telomerase activity. These derivatives showed excellent selectivity for telomeric G-quadruplex DNA over duplex.
Co-reporter:Dong-Fang Liu, Yan-Ping Li, Tian-Miao Ou, Shi-Liang Huang, Lian-Quan Gu, Min Huang, Zhi-Shu Huang
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 15) pp:4237-4240
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmcl.2009.05.103
A series of icariin derivatives were synthesized. Their multidrug resistance (MDR) reversal activities were evaluated by MTT assay and the results indicated that the derivatives were the potent modulators of MDR. It was showed that the derivatives significantly increased the intracellular accumulation of ADR in MCF-7/ADR cells compared with drug sensitive MCF-7 cells. The results of bi-directional assay and reverse transcription polymerase chain reaction (RT-PCR) assay showed that the derivatives had high inhibitory activity against P-gp efflux function and significantly down-regulated on the expression of P-gp.
Co-reporter:Huang Tang, Xiao-Dong Wang, Yong-Biao Wei, Shi-Liang Huang, Zhi-Shu Huang, Jia-Heng Tan, Lin-Kun An, Jian-Yong Wu, Albert Sun-Chi Chan, Lian-Quan Gu
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 5) pp:973-980
Publication Date(Web):May 2008
DOI:10.1016/j.ejmech.2007.07.004
A series of novel oxoisoaporphine alkaloid derivatives, 9-aminoalkanamido-1-azabenzanthrone (general formula Ar–NHCO(CH2)nNR2, Ar = 1-azabenzanthrone, n = 1, 2 or 3), had been synthesized. Compared with 1-azabenzanthrone, the derivatives had significantly higher DNA binding affinity with calf thymus DNA, and higher potent cytotoxicity against different tumor cell lines. The cytotoxicity and the structure–activity relationship of the prepared compounds were studied. The derivatives with two methylene groups (n = 2), and piperidine or ethanolamine functional group in the side chain exhibited highest DNA binding affinity and cytotoxicity.
Co-reporter:Yu-Jing Lu, Tian-Miao Ou, Jia-Heng Tan, Jin-Qiang Hou, Wei-Yan Shao, Dan Peng, Ning Sun, Xiao-Dong Wang, Wei-Bin Wu, Xian-Zhang Bu, Zhi-Shu Huang, Dik-Lung Ma, Kwok-Yin Wong and Lian-Quan Gu
Journal of Medicinal Chemistry 2008 Volume 51(Issue 20) pp:6381-6392
Publication Date(Web):September 27, 2008
DOI:10.1021/jm800497p
A series of 5-N-methyl quindoline (cryptolepine) derivatives (2a−x) as telomeric quadruplex ligands was synthesized and evaluated. The designed ligands possess a positive charge at the 5-N position of the aromatic quindoline scaffold. The quadruplex binding of these compounds was evaluated by circular dichroism (CD) spectroscopy, fluorescence resonance energy transfer (FRET) melting assay, polymerase chain reaction (PCR) stop assay, nuclear magnetic resonance (NMR), and molecular modeling studies. Introduction of a positive charge not only significantly improved the binding ability but also induced the selectivity toward antiparallel quadruplex, whereas the nonmethylated derivatives tended to stabilize hybrid-type quadruplexes. NMR and molecular modeling studies revealed that the ligands stacked on the external G-quartets and the positively charged 5-N atom could contribute to the stabilizing ability. Long-term exposure of human cancer cells to 2r showed a remarkable cessation in population growth and cellular senescence phenotype and accompanied by a shortening of the telomere length.
Co-reporter:Wan-Jin Zhang, Tian-Miao Ou, Yu-Jing Lu, Ying-Yu Huang, Wei-Bin Wu, Zhi-Shu Huang, Jin-Lin Zhou, Kwok-Yin Wong, Lian-Quan Gu
Bioorganic & Medicinal Chemistry 2007 Volume 15(Issue 16) pp:5493-5501
Publication Date(Web):15 August 2007
DOI:10.1016/j.bmc.2007.05.050
The interaction of berberine and its 9-substituted derivatives with human telomeric DNA d[G3(T2AG3)3](telo21) has been investigated via CD spectroscopy, fluorescence spectroscopy, PCR-stop assay, competitive dialysis, and telomerase repeat amplification protocol (TRAP) assay. The results indicated that these semisynthesized compounds could induce and stabilize the formation of anti-parallel G-quadruplex of telomeric DNA in the presence or absence of metal cations. Compared with berberine, the 9-substituted derivatives exhibit stronger binding affinity with G-quadruplex and higher inhibitory activity for telomerase. Introduction of a side chain with proper length of methylene and terminal amino group to the 9-position of berberine would significantly strengthen the binding affinity with G-quadruplex, resulting in increasing inhibitory effects on the amplification of telo21 DNA and on the telomerase activity.
Co-reporter:Huang Tang, Fang-Xian Ning, Yong-Biao Wei, Shi-Liang Huang, Zhi-Shu Huang, Albert Sun-Chi Chan, Lian-Quan Gu
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 13) pp:3765-3768
Publication Date(Web):1 July 2007
DOI:10.1016/j.bmcl.2007.04.015
A series of 9-aminoalkanamido-1-azabenzanthrones derviatives (3a–i Ar–NHCO(CH2)nNR1R2) and their quaternary methiodide salts (4a–g Ar–NHCO(CH2)nN+(CH3)R1R2I−) were designed and synthesized as acetylcholinesterase (AChE) or butyrylcholinesterase (BuChE) inhibitors. The synthetic compounds exhibited high AChE inhibitory activity with IC50 values in the nanomolar range and high selectivity for AChE over BuChE (45- to 1980-fold). The structure–activity relationships (SARs) were discussed.
Co-reporter:Jin-Qiang Hou, Shuo-Bin Chen, Li-Peng Zan, Tian-Miao Ou, Jia-Heng Tan, Leonard G. Luyt and Zhi-Shu Huang
Chemical Communications 2015 - vol. 51(Issue 1) pp:NaN201-201
Publication Date(Web):2014/11/03
DOI:10.1039/C4CC06951J
To efficiently identify small molecules binding to a G-quadruplex structure while avoiding binding to duplex DNA, we performed a multistep structure-based virtual screening by simultaneously taking into account G-quadruplex DNA and duplex DNA. Among the 13 compounds selected, one outstanding ligand shows significant selectivity for G-quadruplex binding as determined using SPR, FRET-based competition and luciferase activity assay.
Co-reporter:Wei-Bin Wu, Shu-Han Chen, Jin-Qiang Hou, Jia-Heng Tan, Tian-Miao Ou, Shi-Liang Huang, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 8) pp:NaN2986-2986
Publication Date(Web):2011/01/28
DOI:10.1039/C0OB00921K
A series of 2-phenyl-benzopyranopyrimidine (PBPP) derivatives with alkylamino side chains were synthesized and found to be a new type of highly selective ligand to bind with telomeric G-quadruplex DNA, and their biological properties were reported for the first time. Their interactions with telomeric G-quadruplex DNA were studied with FRET melting, surface plasmon resonance, CD spectroscopy, and molecular modeling. Our results showed that the disubstituted PBPP derivatives could strongly bind to and effectively stabilize the telomeric G-quadruplex structure, and had significant selectivity for G-quadruplex over duplex DNA. In comparison, the mono substituted derivatives had much less effect on the G-quadruplex, suggesting that the disubstitution of PBPP is essential for its interaction with the G-quadruplex. Furthermore, telomerase inhibition of the PBPP derivatives and their cellular effects were studied, and compound 11b was found to be the most promising compound as a telomerase inhibitor and telomeric G-quadruplex binding ligand for further development for cancer treatment.
Co-reporter:Jin-Qiang Hou, Jia-Heng Tan, Xiao-Xiao Wang, Shuo-Bin Chen, Si-Yuan Huang, Jin-Wu Yan, Shu-Han Chen, Tian-Miao Ou, Hai-Bin Luo, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 18) pp:NaN6436-6436
Publication Date(Web):2011/06/20
DOI:10.1039/C1OB05884C
G-quadruplex structures are a new class of attractive targets for DNA-interactive anticancer agents. The primary building block of this structure is the G-quartet, which is composed of four coplanar guanines and serves as the major binding site for small molecules. NMR studies and molecular dynamics simulations have suggested that the planarity of G-quartet surface has been highly dynamic in solution. To better investigate how the planarity of unfused aromatic ligand impacts on its quadruplex binding properties, a variety of planarity controllable isaindigotone derivatives were designed and synthesized. The interaction of G-quadruplex DNA with these designed ligands was systematically explored using a series of biophysical studies. The FRET-melting, SPR, and CD spectroscopy results showed that reducing the planarity of their unfused aromatic core resulted in their decreased binding affinity and stabilization ability for G-quadruplex. NMR studies also suggested that these compounds could stack on the G-quartet surface. Such results are in parallel with subsequent molecular modeling studies. A detailed binding energy analysis indicated that van der Waals energy (ΔEvdw) and entropy (TΔS) are responsible for their decreased quadruplex binding and stabilization effect. All these results provided insight information about how quadruplex recognition could be controlled by adjusting the planarity of ligands, which shed light on further development of unfused aromatic molecules as optimal G-quadruplex binding ligands.
Co-reporter:Shuo-Bin Chen, Wei-Bin Wu, Ming-Hao Hu, Tian-Miao Ou, Lian-Quan Gu, Jia-Heng Tan and Zhi-Shu Huang
Chemical Communications 2014 - vol. 50(Issue 81) pp:NaN12176-12176
Publication Date(Web):2014/08/21
DOI:10.1039/C4CC05394J
A novel 2,4,5-triaryl-substituted imidazole (IZCM-1) has been found to display distinct and specific fluorescence enhancement upon binding to parallel G-quadruplexes. Such a sensitive and topology-specific probe is able to light up without affecting the topology or thermal stability of the G-quadruplex sample. Thus, these advantages distinguish IZCM-1 from other G-quadruplex probes.
Co-reporter:Jin-Wu Yan, Shuo-Bin Chen, Hui-Yun Liu, Wen-Jie Ye, Tian-Miao Ou, Jia-Heng Tan, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Chemical Communications 2014 - vol. 50(Issue 52) pp:NaN6930-6930
Publication Date(Web):2014/05/07
DOI:10.1039/C4CC01472C
A tailor-made colorimetric and red-emitting fluorescent dual probe for G-quadruplex nucleic acids was developed by incorporating a coumarin–hemicyanine fluorophore into an isaindigotone framework. The significant and distinct changes in both the color and fluorescence of this probe enable the label-free and visual detection of G-quadruplex structures.
Co-reporter:Shi-Tian Zhuo, Chun-Yan Li, Ming-Hao Hu, Shuo-Bin Chen, Pei-Fen Yao, Shi-Liang Huang, Tian-Miao Ou, Jia-Heng Tan, Lin-Kun An, Ding Li, Lian-Quan Gu and Zhi-Shu Huang
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 24) pp:NaN4005-4005
Publication Date(Web):2013/04/04
DOI:10.1039/C3OB40325D
Topoisomerases (Topo I and Topo II) are very important players in DNA replication, repair, and transcription, and are a promising class of antitumor target. In present study, a series of benzo[a]phenazine derivatives with alkylamino side chains at C-5 were designed, synthesized, and their biological activities were evaluated. Most of derivatives showed good antiproliferative activity with a range of IC50 values of 1–10 μM on the four cancer cell lines HeLa, A549, MCF-7, and HL-60. Topoisomerase-mediated DNA relaxation assay results showed that derivatives could effectively inhibit the activity of both Topo I and Topo II, and the structure–activity relationship studies indicated the importance of introducing an alkylamino side chain. Further mechanism studies revealed that the compounds could stabilize the Topo I–DNA cleavage complexes and inhibit the ATPase activity of hTopo II, indicating that they are a rare class of dual topoisomerase inhibitors by acting as Topo I poisons and Topo II catalytic inhibitors. Moreover, flow cytometric analysis and caspase-3/7 activation assay showed that this class of compounds could induce apoptosis of HL-60 cells.
Co-reporter:Jia-Qiang Wu, Zhi-Ping Qiu, Shang-Shi Zhang, Jing-Gong Liu, Ye-Xing Lao, Lian-Quan Gu, Zhi-Shu Huang, Juan Li and Honggen Wang
Chemical Communications 2015 - vol. 51(Issue 1) pp:NaN80-80
Publication Date(Web):2014/11/04
DOI:10.1039/C4CC07839J
Succession of C–H activation and C–C activation was achieved by using a single rhodium(III) catalyst. Vinylcyclopropanes were used as versatile coupling partners. Mechanistic studies suggest that the olefin insertion step is rate-determining and a facile β-carbon elimination is involved, which represents a novel ring opening mode of vinylcyclopropanes.

















































![Pyrrolo[2,1-b]quinazolin-9(1H)-one, 6,7-difluoro-2,3-dihydro-](/data/chemimg/2702800/642491-85-8.png)
![Pyrrolo[2,1-b]quinazolin-9(1H)-one, 6,7-difluoro-2,3-dihydro-](/data/chemimg/2702800/642491-85-8_b.png)
![1,2-Benzenediamine, N-[2-(4-methyl-1-piperazinyl)ethyl]-](http://img.cochemist.com/ccimg/600800/600725-24-4.png)
![1,2-Benzenediamine, N-[2-(4-methyl-1-piperazinyl)ethyl]-](http://img.cochemist.com/ccimg/600800/600725-24-4_b.png)
![1,2-BENZENEDIAMINE, N-[4-(DIMETHYLAMINO)BUTYL]-](http://img.cochemist.com/ccimg/600800/600725-11-9.png)
![1,2-BENZENEDIAMINE, N-[4-(DIMETHYLAMINO)BUTYL]-](http://img.cochemist.com/ccimg/600800/600725-11-9_b.png)










![SODIUM (2S)-5-AMINO-5-OXO-2-[(PHENYLACETYL)AMINO]PENTANOATE PHENY<WBR />LACETATE (2:1:1)](http://img.cochemist.com/ccimg/303300/303224-88-6.png)
![SODIUM (2S)-5-AMINO-5-OXO-2-[(PHENYLACETYL)AMINO]PENTANOATE PHENY<WBR />LACETATE (2:1:1)](http://img.cochemist.com/ccimg/303300/303224-88-6_b.png)


![2-Penten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-, (2E)-](http://img.cochemist.com/ccimg/199000/198977-95-6.png)
![2-Penten-1-one, 5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-phenyl-, (2E)-](http://img.cochemist.com/ccimg/199000/198977-95-6_b.png)



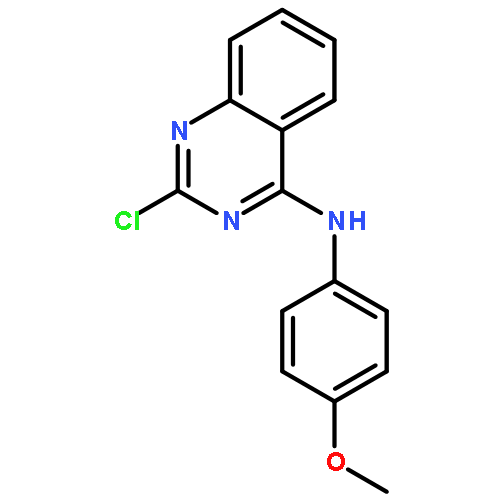
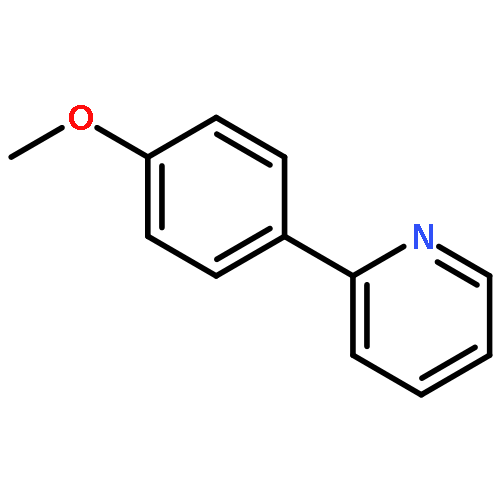
![Quinoline, 2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](/data/chemimg/3761900/190437-90-2.png)
![Quinoline, 2-[(1E)-2-(4-methoxyphenyl)ethenyl]-](/data/chemimg/3761900/190437-90-2_b.png)
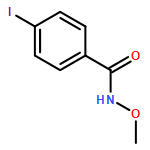
![[1,1'-Biphenyl]-4-carboxamide, N-methoxy-](/data/chemimg/105400/173171-19-2.png)
![[1,1'-Biphenyl]-4-carboxamide, N-methoxy-](/data/chemimg/105400/173171-19-2_b.png)








![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2.png)
![Benzoic acid, 4-[(methoxyamino)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/137100/137042-75-2_b.png)
![Indolizino[2,3-g]quinoline-6-carboxylic acid, 5,12-dihydro-5,12-dioxo-,ethyl ester](http://img.cochemist.com/ccimg/134800/134793-78-5.png)
![Indolizino[2,3-g]quinoline-6-carboxylic acid, 5,12-dihydro-5,12-dioxo-,ethyl ester](http://img.cochemist.com/ccimg/134800/134793-78-5_b.png)






![Phosphonium, [2-(4-fluorophenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/120400/120302-61-6.png)
![Phosphonium, [2-(4-fluorophenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/120400/120302-61-6_b.png)



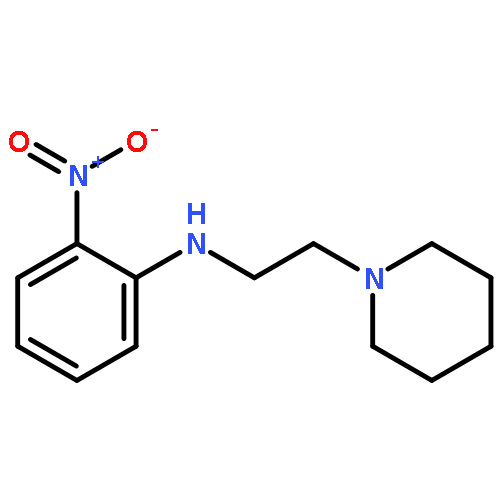







![1,2-Benzenediamine, N-[3-(diethylamino)propyl]-](http://img.cochemist.com/ccimg/92000/91971-96-9.png)
![1,2-Benzenediamine, N-[3-(diethylamino)propyl]-](http://img.cochemist.com/ccimg/92000/91971-96-9_b.png)






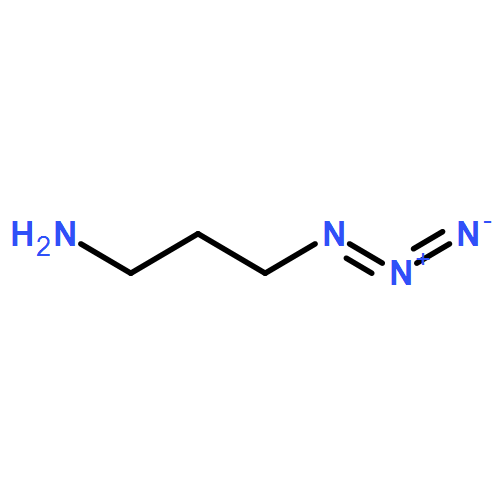


![Phenanthro[1,2-b]furan-10,11-dione, 1,2-dihydro-1,6-dimethyl-, (1R)-](/data/chemimg/1469400/87205-99-0.png)
![Phenanthro[1,2-b]furan-10,11-dione, 1,2-dihydro-1,6-dimethyl-, (1R)-](/data/chemimg/1469400/87205-99-0_b.png)






![Quinoline, 2-[(1E)-2-(2-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/77700/77669-18-2.png)
![Quinoline, 2-[(1E)-2-(2-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/77700/77669-18-2_b.png)
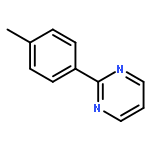
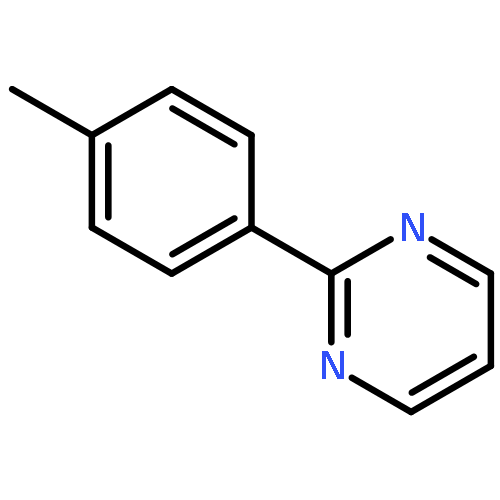








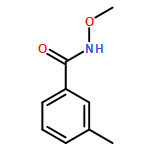
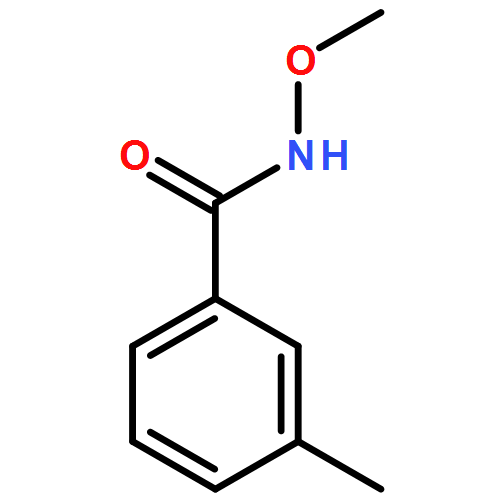
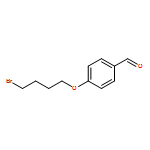

![Benzofuro[3,2-b]quinoline, 11-chloro-](/data/chemimg/2543200/69764-19-8.png)
![Benzofuro[3,2-b]quinoline, 11-chloro-](/data/chemimg/2543200/69764-19-8_b.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-2-nitro-](/data/chemimg/1917700/67718-36-9.png)
![Benzamide, N-[2-(aminocarbonyl)phenyl]-2-nitro-](/data/chemimg/1917700/67718-36-9_b.png)


![Phosphonium, [2-(4-iodophenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/67800/67717-17-3.png)
![Phosphonium, [2-(4-iodophenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/67800/67717-17-3_b.png)








![1,2-Benzenediamine, N-[3-(4-morpholinyl)propyl]-](http://img.cochemist.com/ccimg/62600/62553-49-5.png)
![1,2-Benzenediamine, N-[3-(4-morpholinyl)propyl]-](http://img.cochemist.com/ccimg/62600/62553-49-5_b.png)
![Benzo[h]quinoline, 4-chloro-2-methyl-](http://img.cochemist.com/ccimg/61800/61773-04-4.png)
![Benzo[h]quinoline, 4-chloro-2-methyl-](http://img.cochemist.com/ccimg/61800/61773-04-4_b.png)
![4,9,9-trimethyl-9,10,11,12-tetrahydrofuro[3,2-c]naphtho[2,1-e]oxepine-1,3-dione](http://img.cochemist.com/ccimg/61100/61077-78-9.png)
![4,9,9-trimethyl-9,10,11,12-tetrahydrofuro[3,2-c]naphtho[2,1-e]oxepine-1,3-dione](http://img.cochemist.com/ccimg/61100/61077-78-9_b.png)






![Phosphonium, [2-oxo-2-[4-(trifluoromethyl)phenyl]ethyl]triphenyl-, bromide (1:1)](/data/chemimg/1852200/56893-13-1.png)
![Phosphonium, [2-oxo-2-[4-(trifluoromethyl)phenyl]ethyl]triphenyl-, bromide (1:1)](/data/chemimg/1852200/56893-13-1_b.png)


![Ethanone, 1-[4-(trifluoromethyl)phenyl]-2-(triphenylphosphoranylidene)-](/data/chemimg/3765500/56893-05-1.png)
![Ethanone, 1-[4-(trifluoromethyl)phenyl]-2-(triphenylphosphoranylidene)-](/data/chemimg/3765500/56893-05-1_b.png)
![1,2-BENZENEDIAMINE, 4-CHLORO-N1-[3-(DIMETHYLAMINO)PROPYL]-](http://img.cochemist.com/ccimg/56800/56756-22-0.png)
![1,2-BENZENEDIAMINE, 4-CHLORO-N1-[3-(DIMETHYLAMINO)PROPYL]-](http://img.cochemist.com/ccimg/56800/56756-22-0_b.png)






![N-[1-(4-METHYLPHENYL)ETHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/54600/54582-23-9.png)
![N-[1-(4-METHYLPHENYL)ETHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/54600/54582-23-9_b.png)
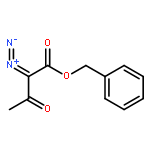
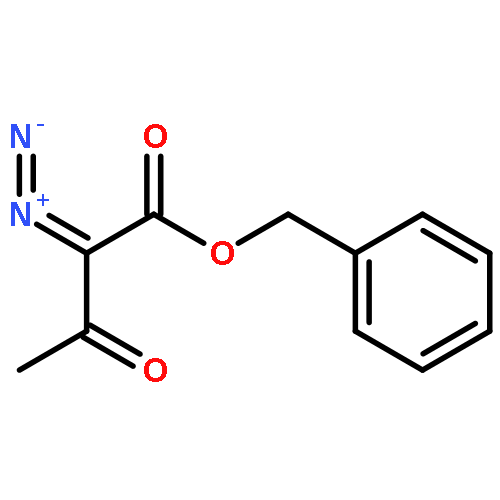


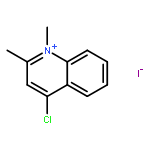

![BENZENAMINE, N,N-DIMETHYL-4-[(1E)-2-(2-QUINOLINYL)ETHENYL]-](http://img.cochemist.com/ccimg/48200/48189-64-6.png)
![BENZENAMINE, N,N-DIMETHYL-4-[(1E)-2-(2-QUINOLINYL)ETHENYL]-](http://img.cochemist.com/ccimg/48200/48189-64-6_b.png)
![9-chloro-2,3-dihydro-1H-cyclopenta[b]quinoline](http://img.cochemist.com/ccimg/40600/40528-00-5.png)
![9-chloro-2,3-dihydro-1H-cyclopenta[b]quinoline](http://img.cochemist.com/ccimg/40600/40528-00-5_b.png)










![Quinoline, 2-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1815300/38101-93-8.png)
![Quinoline, 2-[(1E)-2-(4-nitrophenyl)ethenyl]-](/data/chemimg/1815300/38101-93-8_b.png)



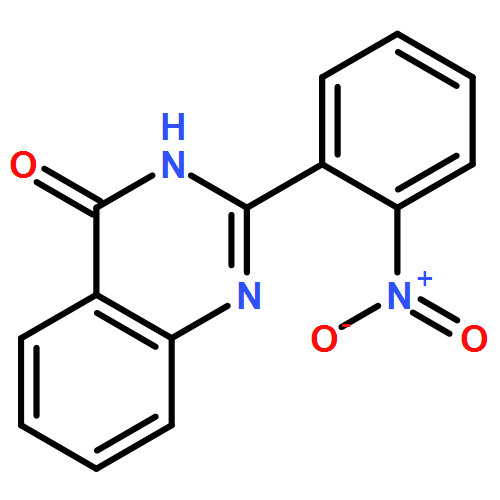


![9H-Imidazo[1,2-a]benzimidazole-9-ethanamine, N,N-diethyl-2-phenyl-](http://img.cochemist.com/ccimg/33800/33729-71-4.png)
![9H-Imidazo[1,2-a]benzimidazole-9-ethanamine, N,N-diethyl-2-phenyl-](http://img.cochemist.com/ccimg/33800/33729-71-4_b.png)
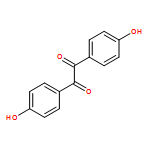





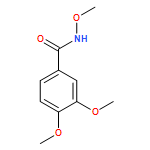














![2-N-[3-(DIMETHYLAMINO)PROPYL]BENZENE-1,2-DIAMINE](http://img.cochemist.com/ccimg/21700/21627-59-8.png)
![2-N-[3-(DIMETHYLAMINO)PROPYL]BENZENE-1,2-DIAMINE](http://img.cochemist.com/ccimg/21700/21627-59-8_b.png)

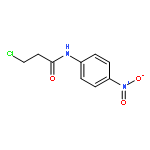

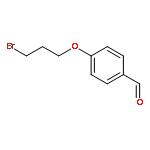
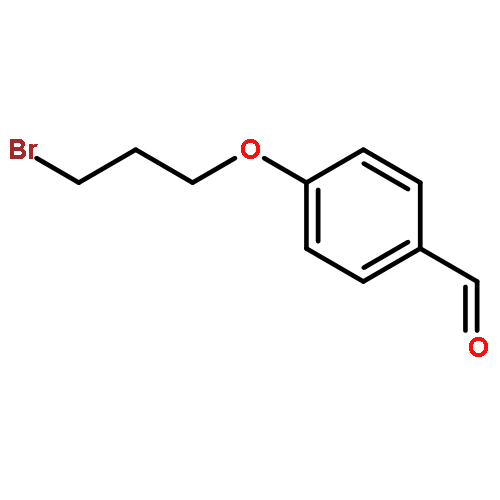


![2-(Benzo[d]oxazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/15400/15344-56-6.png)
![2-(Benzo[d]oxazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/15400/15344-56-6_b.png)
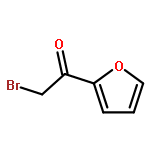

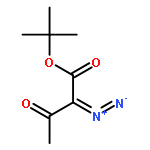
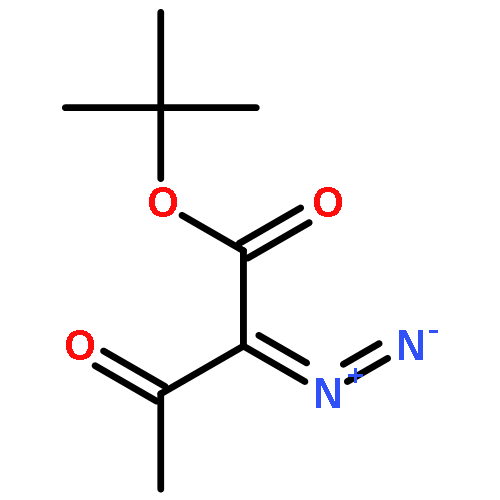
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)
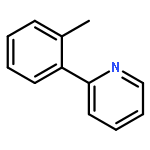
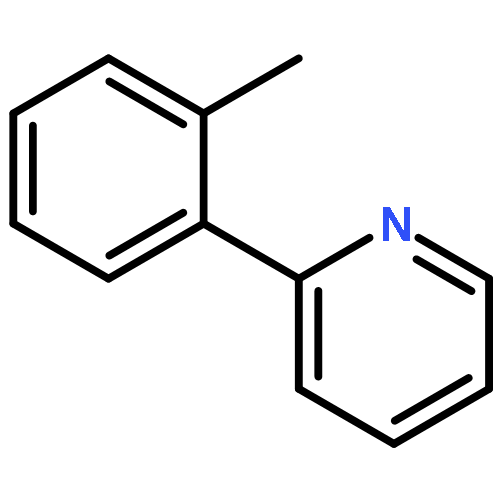












![11-chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinoline](http://img.cochemist.com/ccimg/5800/5778-71-2.png)
![11-chloro-7,8,9,10-tetrahydro-6H-cyclohepta[b]quinoline](http://img.cochemist.com/ccimg/5800/5778-71-2_b.png)




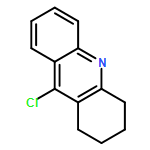
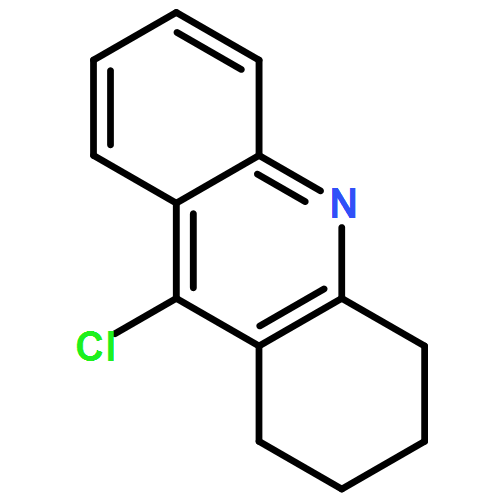

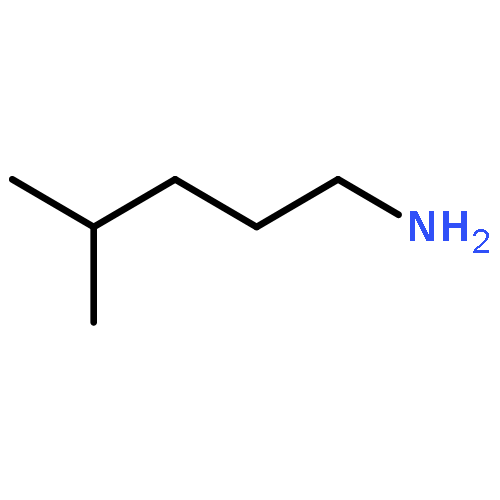




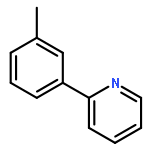




phosphonium](http://img.cochemist.com/ccimg/3000/2928-58-7.png)
phosphonium](http://img.cochemist.com/ccimg/3000/2928-58-7_b.png)
![2-(1-Methyl-1H-benzo[d]imidazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/2800/2735-62-8.png)
![2-(1-Methyl-1H-benzo[d]imidazol-2-yl)acetonitrile](http://img.cochemist.com/ccimg/2800/2735-62-8_b.png)
![Phosphonium, [2-(4-chlorophenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/2700/2689-81-8.png)
![Phosphonium, [2-(4-chlorophenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/2700/2689-81-8_b.png)
![Phosphonium, [2-(4-methylphenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/2700/2689-61-4.png)
![Phosphonium, [2-(4-methylphenyl)-2-oxoethyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/2700/2689-61-4_b.png)









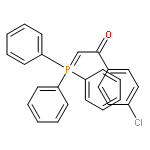

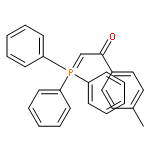
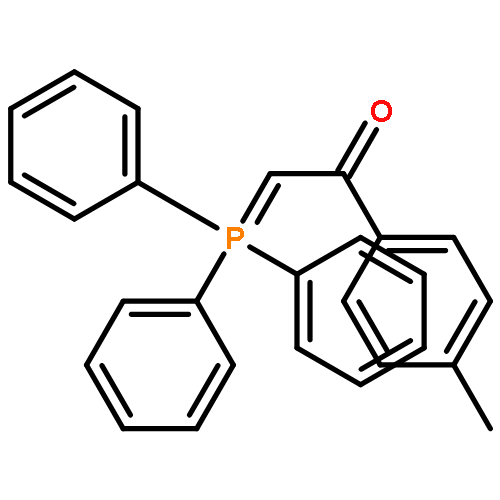






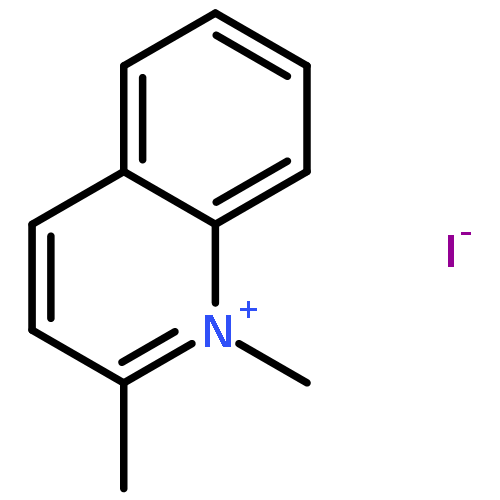




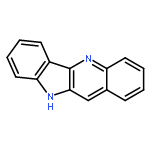

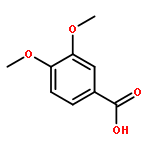
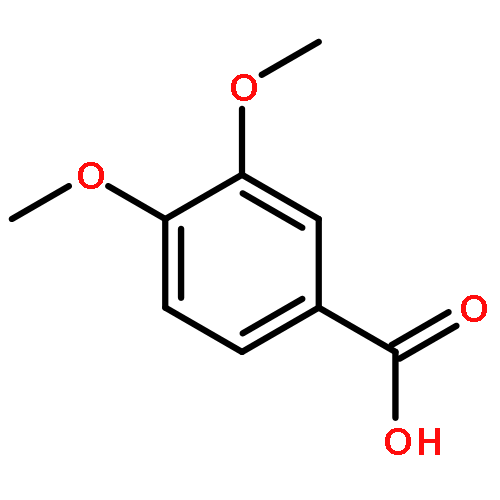




![Ethanone, 1-[4-(2-pyridinyl)phenyl]-](http://img.cochemist.com/ccimg/173700/173681-56-6.png)
![Ethanone, 1-[4-(2-pyridinyl)phenyl]-](http://img.cochemist.com/ccimg/173700/173681-56-6_b.png)
![4(1H)-Quinazolinone, 2-[1-(phenylhydrazono)ethyl]-](http://img.cochemist.com/ccimg/156000/155912-14-4.png)
![4(1H)-Quinazolinone, 2-[1-(phenylhydrazono)ethyl]-](http://img.cochemist.com/ccimg/156000/155912-14-4_b.png)