Co-reporter:Yanfa Yan;Zewen Xiao;Weiwei Meng
The Journal of Physical Chemistry Letters October 6, 2016 Volume 7(Issue 19) pp:3903-3907
Publication Date(Web):September 16, 2016
DOI:10.1021/acs.jpclett.6b01834
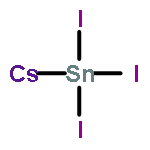
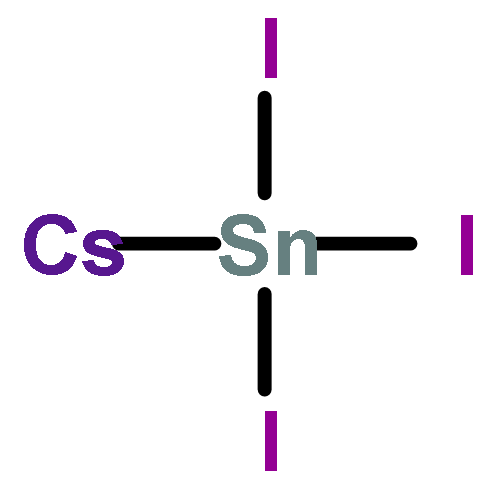
Synthesis of cubic-phase AgBi2I7 iodobismuthate thin films and fabrication of air-stable Pb-free solar cells using the AgBi2I7 absorber have recently been reported. On the basis of X-ray diffraction (XRD) analysis and nominal composition, it was suggested that the synthesized films have a cubic ThZr2H7 crystal structure with AgBi2I7 stoichiometry. Through careful examination of the proposed structure and computational evaluation of the phase stability and bandgap, we find that the reported “AgBi2I7” films cannot be forming with the ThZr2H7-type structure, but rather more likely adopt an Ag-deficient AgBiI4 type. Both the experimental X-ray diffraction pattern and bandgap can be better explained by the AgBiI4 structure. Additionally, the proposed AgBiI4 structure, with octahedral bismuth coordination, removes unphysically short Bi–I bonding within the [BiI8] hexahedra of the ThZr2I7 model. Our results provide critical insights for assessing the photovoltaic properties of AgBi2I7 iodobismuthate materials.
Co-reporter:Ke-zhao Du, Xiaoming Wang, Qiwei Han, Yanfa Yan, and David B. Mitzi
ACS Energy Letters October 13, 2017 Volume 2(Issue 10) pp:2486-2486
Publication Date(Web):September 26, 2017
DOI:10.1021/acsenergylett.7b00824
Compositional engineering, which can enrich the database of prospective materials and offer new or enhanced properties, represents one of the key focal points within halide perovskite research. Compositional engineering studies often focus on A+ and X– site substitutions, within the ABX3 perovskite structure, due to the relative ease of varying these sites. However, alloying on the B site can play a more important role in generating novel properties and decreasing Pb toxicity for Pb-based systems. To date, B site substitution has primarily been confined to single-element alloying. Herein, a heterovalent co-alloying strategy for the B site of halide perovskites is proposed. AgIBiIII and AgISbIII are co-alloyed into a host crystal of APbBr3 (A = Cs and methylammonium), leading to a larger range of prospective alloying elements on the perovskite B site. Density functional theory-based first-principles calculations provide a possible rational for the red shift of the bandgap and blue shift of the photoluminescence (PL) in the alloying experiments.
Co-reporter:Zewen Xiao, Ke-Zhao Du, Weiwei Meng, Jianbo Wang, David B. Mitzi, and Yanfa Yan
Journal of the American Chemical Society May 3, 2017 Volume 139(Issue 17) pp:6054-6054
Publication Date(Web):April 18, 2017
DOI:10.1021/jacs.7b02227
Recently, there has been substantial interest in developing double-B-cation halide perovskites, which hold the potential to overcome the toxicity and instability issues inherent within emerging lead halide-based solar absorber materials. Among all double perovskites investigated, In(I)-based Cs2InBiCl6 and Cs2InSbCl6 have been proposed as promising thin-film photovoltaic absorber candidates, with computational examination predicting suitable materials properties, including direct bandgap and small effective masses for both electrons and holes. In this study, we report the intrinsic instability of Cs2In(I)M(III)X6 (M = Bi, Sb; X = halogen) double perovskites by a combination of density functional theory and experimental study. Our results suggest that the In(I)-based double perovskites are unstable against oxidation into In(III)-based compounds. Further, the results show the need to consider reduction–oxidation (redox) chemistry when predicting stability of new prospective electronic materials, especially when less common oxidation states are involved.
Co-reporter:Weiwei Meng, Xiaoming Wang, Zewen Xiao, Jianbo Wang, David B. Mitzi, and Yanfa Yan
The Journal of Physical Chemistry Letters July 6, 2017 Volume 8(Issue 13) pp:2999-2999
Publication Date(Web):June 12, 2017
DOI:10.1021/acs.jpclett.7b01042
Using density functional theory calculations, we analyze the optical absorption properties of lead (Pb)-free metal halide perovskites (AB2+X3) and double perovskites (A2B+B3+X6) (A = Cs or monovalent organic ion, B2+ = non-Pb divalent metal, B+ = monovalent metal, B3+ = trivalent metal, X = halogen). We show that if B2+ is not Sn or Ge, Pb-free metal halide perovskites exhibit poor optical absorptions because of their indirect band gap nature. Among the nine possible types of Pb-free metal halide double perovskites, six have direct band gaps. Of these six types, four show inversion symmetry-induced parity-forbidden or weak transitions between band edges, making them not ideal for thin-film solar cell applications. Only one type of Pb-free double perovskite shows optical absorption and electronic properties suitable for solar cell applications, namely, those with B+ = In, Tl and B3+ = Sb, Bi. Our results provide important insights for designing new metal halide perovskites and double perovskites for optoelectronic applications.
Co-reporter:Ke-zhao Du, Qing Tu, Xu Zhang, Qiwei Han, Jie Liu, Stefan Zauscher, and David B. Mitzi
Inorganic Chemistry August 7, 2017 Volume 56(Issue 15) pp:9291-9291
Publication Date(Web):July 27, 2017
DOI:10.1021/acs.inorgchem.7b01094
A series of two-dimensional (2D) hybrid organic–inorganic perovskite (HOIP) crystals, based on acene alkylamine cations (i.e., phenylmethylammonium (PMA), 2-phenylethylammonium (PEA), 1-(2-naphthyl)methanammonium (NMA), and 2-(2-naphthyl)ethanammonium (NEA)) and lead(II) halide (i.e., PbX42–, X = Cl, Br, and I) frameworks, and their corresponding thin films were fabricated and examined for structure–property relationship. Several new or redetermined crystal structures are reported, including those for (NEA)2PbI4, (NEA)2PbBr4, (NMA)2PbBr4, (PMA)2PbBr4, and (PEA)2PbI4. Non-centrosymmetric structures from among these 2D HOIPs were confirmed by piezoresponse force microscopy—especially noteworthy is the structure of (PMA)2PbBr4, which was previously reported as centrosymmetric. Examination of the impact of organic cation and inorganic layer choice on the exciton absorption/emission properties, among the set of compounds considered, reveals that perovskite layer distortion (i.e., Pb–I–Pb bond angle between adjacent PbI6 octahedra) has a more global effect on the exciton properties than octahedral distortion (i.e., variation of I–Pb–I bond angles and discrepancy among Pb–I bond lengths within each PbI6 octahedron). In addition to the characteristic sharp exciton emission for each perovskite, (PMA)2PbCl4, (PEA)2PbCl4, (NMA)2PbCl4, and (PMA)2PbBr4 exhibit separate, broad “white” emission in the long wavelength range. Piezoelectric compounds identified from these 2D HOIPs may be considered for future piezoresponse-type energy or electronic applications.
Co-reporter:Zewen Xiao;Weiwei Meng;Jianbo Wang;Yanfa Yan
Materials Horizons (2014-Present) 2017 vol. 4(Issue 2) pp:206-216
Publication Date(Web):2017/03/06
DOI:10.1039/C6MH00519E
Searching for promising nontoxic and air-stable perovskite absorbers for solar cell applications has drawn extensive attention. Here, we show that a promising perovskite absorber should exhibit a high electronic dimensionality. Semiconductors that exhibit a high structural dimensionality, but a low electronic dimensionality have less promise as an absorber, because of barriers to isotropic current flow, enhanced electron/hole effective masses and fundamentally deeper defect states (more effective at causing recombination). Our concept accounts for the device performance of the perovskite-based solar cells reported in literature so far.
Co-reporter:Dr. Ke-zhao Du;Dr. Weiwei Meng;Dr. Xiaoming Wang; Yanfa Yan; David B. Mitzi
Angewandte Chemie International Edition 2017 Volume 56(Issue 28) pp:8158-8162
Publication Date(Web):2017/07/03
DOI:10.1002/anie.201703970
AbstractThe double perovskite family, A2MIMIIIX6, is a promising route to overcome the lead toxicity issue confronting the current photovoltaic (PV) standout, CH3NH3PbI3. Given the generally large indirect band gap within most known double perovskites, band-gap engineering provides an important approach for targeting outstanding PV performance within this family. Using Cs2AgBiBr6 as host, band-gap engineering through alloying of InIII/SbIII has been demonstrated in the current work. Cs2Ag(Bi1−xMx)Br6 (M=In, Sb) accommodates up to 75 % InIII with increased band gap, and up to 37.5 % SbIII with reduced band gap; that is, enabling ca. 0.41 eV band gap modulation through introduction of the two metals, with smallest value of 1.86 eV for Cs2Ag(Bi0.625Sb0.375)Br6. Band structure calculations indicate that opposite band gap shift directions associated with Sb/In substitution arise from different atomic configurations for these atoms. Associated photoluminescence and environmental stability of the three-metal systems are also assessed.
Co-reporter:Dr. Zewen Xiao;Dr. Ke-Zhao Du;Weiwei Meng; David B. Mitzi; Yanfa Yan
Angewandte Chemie 2017 Volume 129(Issue 40) pp:12275-12279
Publication Date(Web):2017/09/25
DOI:10.1002/ange.201705113
AbstractRecently, CuI- and AgI-based halide double perovskites have been proposed as promising candidates for overcoming the toxicity and instability issues inherent within the emerging Pb-based halide perovskite absorbers. However, up to date, only AgI-based halide double perovskites have been experimentally synthesized; there are no reports on successful synthesis of CuI-based analogues. Here we show that, owing to the much higher energy level for the Cu 3d10 orbitals than for the Ag 4d10 orbitals, CuI atoms energetically favor 4-fold coordination, forming [CuX4] tetrahedra (X=halogen), but not 6-fold coordination as required for [CuX6] octahedra. In contrast, AgI atoms can have both 6- and 4-fold coordinations. Our density functional theory calculations reveal that the synthesis of CuI halide double perovskites may instead lead to non-perovskites containing [CuX4] tetrahedra, as confirmed by our material synthesis efforts.
Co-reporter:Donghyeop Shin;Bayrammurad Saparov
Advanced Energy Materials 2017 Volume 7(Issue 11) pp:
Publication Date(Web):2017/06/01
DOI:10.1002/aenm.201602366
Application of zinc-blende-related chalcogenide absorbers such as CdTe and Cu(In,Ga)Se2 (CIGSe) has enabled remarkable advancement in laboratory- and commercial-scale thin-film photovoltaic performance; however concerns remain regarding the toxicity (CdTe) and scarcity (CIGSe/CdTe) of the constituent elements. Recently, kesterite-based Cu2ZnSn(S,Se)4 (CZTSSe) materials have emerged as attractive non-toxic and earth-abundant absorber candidates. Despite the similarities between CZTSSe and CIGSe/CdTe, the record power conversion efficiency of CZTSSe solar cells (12.6%) remains significantly lower than that of CIGSe (22.6%) and CdTe (22.1%) devices, with the performance gap primarily being attributed to cationic disordering and associated band tailing. To capture the promise of kesterite-like materials as prospective “drop-in” earth-abundant replacements for closely-related CIGSe, current research has focused on several key directions to control disorder, including: (i) examination of the interaction between processing conditions and atomic site disorder, (ii) isoelectronic cation substitution to introduce ionic size mismatch, and (iii) structural diversification beyond the zinc-blende-type coordination environment. In this review, recent efforts targeting accurate identification and engineering of anti-site disorder in kesterite-based CZTSSe are considered. Lessons learned from CZTSSe are applied to other complex chalcogenide semiconductors, in an effort to develop promising pathways to avoid anti-site disordering and associated band tailing in future high-performance earth-abundant photovoltaic technologies.
Co-reporter:Donghyeop Shin;Tong Zhu;Xuan Huang;Oki Gunawan;Volker Blum
Advanced Materials 2017 Volume 29(Issue 24) pp:
Publication Date(Web):2017/06/01
DOI:10.1002/adma.201606945
In recent years, Cu2ZnSn(S,Se)4 (CZTSSe) materials have enabled important progress in associated thin-film photovoltaic (PV) technology, while avoiding scarce and/or toxic metals; however, cationic disorder and associated band tailing fundamentally limit device performance. Cu2BaSnS4 (CBTS) has recently been proposed as a prospective alternative large bandgap (~2 eV), environmentally friendly PV material, with ~2% power conversion efficiency (PCE) already demonstrated in corresponding devices. In this study, a two-step process (i.e., precursor sputter deposition followed by successive sulfurization/selenization) yields high-quality nominally pinhole-free films with large (>1 µm) grains of selenium-incorporated (x = 3) Cu2BaSnS4−xSex (CBTSSe) for high-efficiency PV devices. By incorporating Se in the sulfide film, absorber layers with 1.55 eV bandgap, ideal for single-junction PV, have been achieved within the CBTSSe trigonal structural family. The abrupt transition in quantum efficiency data for wavelengths above the absorption edge, coupled with a strong sharp photoluminescence feature, confirms the relative absence of band tailing in CBTSSe compared to CZTSSe. For the first time, by combining bandgap tuning with an air-annealing step, a CBTSSe-based PV device with 5.2% PCE (total area 0.425 cm2) is reported, >2.5× better than the previous champion pure sulfide device. These results suggest substantial promise for the emerging Se-rich Cu2BaSnS4–xSex family for high-efficiency and earth-abundant PV.
Co-reporter:Dr. Ke-zhao Du;Dr. Weiwei Meng;Dr. Xiaoming Wang; Yanfa Yan; David B. Mitzi
Angewandte Chemie 2017 Volume 129(Issue 28) pp:8270-8274
Publication Date(Web):2017/07/03
DOI:10.1002/ange.201703970
AbstractThe double perovskite family, A2MIMIIIX6, is a promising route to overcome the lead toxicity issue confronting the current photovoltaic (PV) standout, CH3NH3PbI3. Given the generally large indirect band gap within most known double perovskites, band-gap engineering provides an important approach for targeting outstanding PV performance within this family. Using Cs2AgBiBr6 as host, band-gap engineering through alloying of InIII/SbIII has been demonstrated in the current work. Cs2Ag(Bi1−xMx)Br6 (M=In, Sb) accommodates up to 75 % InIII with increased band gap, and up to 37.5 % SbIII with reduced band gap; that is, enabling ca. 0.41 eV band gap modulation through introduction of the two metals, with smallest value of 1.86 eV for Cs2Ag(Bi0.625Sb0.375)Br6. Band structure calculations indicate that opposite band gap shift directions associated with Sb/In substitution arise from different atomic configurations for these atoms. Associated photoluminescence and environmental stability of the three-metal systems are also assessed.
Co-reporter:Dr. Zewen Xiao;Dr. Ke-Zhao Du;Weiwei Meng; David B. Mitzi; Yanfa Yan
Angewandte Chemie International Edition 2017 Volume 56(Issue 40) pp:12107-12111
Publication Date(Web):2017/09/25
DOI:10.1002/anie.201705113
AbstractRecently, CuI- and AgI-based halide double perovskites have been proposed as promising candidates for overcoming the toxicity and instability issues inherent within the emerging Pb-based halide perovskite absorbers. However, up to date, only AgI-based halide double perovskites have been experimentally synthesized; there are no reports on successful synthesis of CuI-based analogues. Here we show that, owing to the much higher energy level for the Cu 3d10 orbitals than for the Ag 4d10 orbitals, CuI atoms energetically favor 4-fold coordination, forming [CuX4] tetrahedra (X=halogen), but not 6-fold coordination as required for [CuX6] octahedra. In contrast, AgI atoms can have both 6- and 4-fold coordinations. Our density functional theory calculations reveal that the synthesis of CuI halide double perovskites may instead lead to non-perovskites containing [CuX4] tetrahedra, as confirmed by our material synthesis efforts.
Co-reporter:Bayrammurad Saparov and David B. Mitzi
Chemical Reviews 2016 Volume 116(Issue 7) pp:4558
Publication Date(Web):April 4, 2016
DOI:10.1021/acs.chemrev.5b00715
Although known since the late 19th century, organic–inorganic perovskites have recently received extraordinary research community attention because of their unique physical properties, which make them promising candidates for application in photovoltaic (PV) and related optoelectronic devices. This review will explore beyond the current focus on three-dimensional (3-D) lead(II) halide perovskites, to highlight the great chemical flexibility and outstanding potential of the broader class of 3-D and lower dimensional organic-based perovskite family for electronic, optical, and energy-based applications as well as fundamental research. The concept of a multifunctional organic–inorganic hybrid, in which the organic and inorganic structural components provide intentional, unique, and hopefully synergistic features to the compound, represents an important contemporary target.
Co-reporter:Weijun Ke;Chuanxiao Xiao;Changlei Wang;Bayrammurad Saparov;Hsin-Sheng Duan;Dewei Zhao;Zewen Xiao;Philip Schulz;Steven P. Harvey;Weiqiang Liao;Weiwei Meng;Yue Yu;Alexer J. Cimaroli;Chun-Sheng Jiang;Kai Zhu;Mowafak Al-Jassim;Guojia Fang;Yanfa Yan
Advanced Materials 2016 Volume 28( Issue 26) pp:5214-5221
Publication Date(Web):
DOI:10.1002/adma.201600594
Co-reporter:Weiwei Meng, Bayrammurad Saparov, Feng Hong, Jianbo Wang, David B. Mitzi, and Yanfa Yan
Chemistry of Materials 2016 Volume 28(Issue 3) pp:821
Publication Date(Web):January 11, 2016
DOI:10.1021/acs.chemmater.5b04213
Through density functional theory calculations, we show that the alloy perovskite system BaZr1–xTixS3 (x < 0.25) is a promising candidate for producing high power conversion efficiency (PCE) solar cells with ultrathin absorber layers. To maximize the minority carrier lifetime, which is important for achieving high PCE, the defect calculations show that BaZr1–xTixS3 films should be synthesized under moderate (i.e., near stoichiometric) growth conditions to minimize the formation of deep-level defects. The perovskite BaZrS3 is also found to exhibit ambipolar self-doping properties, indicating the ability to form homo p–n junctions. However, our theoretical calculations and experimental solid-state reaction efforts indicate that the doped perovskite BaZr1–xTixS3 (x > 0) may not be stable under thermal equilibrium growth conditions. Calculations of decomposition energies suggest that introducing compressive strain may be a plausible approach to stabilize BaZr1–xTixS3 thin films.
Co-reporter:Bayrammurad Saparov, Jon-Paul Sun, Weiwei Meng, Zewen Xiao, Hsin-Sheng Duan, Oki Gunawan, Donghyeop Shin, Ian G. Hill, Yanfa Yan, and David B. Mitzi
Chemistry of Materials 2016 Volume 28(Issue 7) pp:2315
Publication Date(Web):March 13, 2016
DOI:10.1021/acs.chemmater.6b00433
In this work, we describe details of a two-step deposition approach that enables the preparation of continuous and well-structured thin films of Cs2SnI6, which is a one-half Sn-deficient 0-D perovskite derivative (i.e., the compound can also be written as CsSn0.5I3, with a structure consisting of isolated SnI64– octahedra). The films were characterized using powder X-ray diffraction (PXRD), scanning electron microscopy (SEM), thermogravimetric analysis (TGA), UV–vis spectroscopy, photoluminescence (PL), photoelectron spectroscopy (UPS, IPES, XPS), and Hall effect measurements. UV–vis and PL measurements indicate that the obtained Cs2SnI6 film is a semiconductor with a band gap of 1.6 eV. This band gap was further confirmed by the UPS and IPES spectra, which were well reproduced by the calculated density of states with the HSE hybrid functional. The Cs2SnI6 films exhibited n-type conduction with a carrier density of 6(1) × 1016 cm–3 and mobility of 2.9(3) cm2/V·s. While the computationally derived band structure for Cs2SnI6 shows significant dispersion along several directions in the Brillouin zone near the band edges, the valence band is relatively flat along the Γ–X direction, indicative of a more limited hole minority carrier mobility compared to analogous values for the electrons. The ionization potential (IP) and electron affinity (EA) were determined to be 6.4 and 4.8 eV, respectively. The Cs2SnI6 films show some enhanced stability under ambient air, compared to methylammonium lead(II) iodide perovskite films stored under similar conditions; however, the films do decompose slowly, yielding a CsI impurity. These findings are discussed in the context of suitability of Cs2SnI6 for photovoltaic and related optoelectronic applications.
Co-reporter:Donghyeop Shin, Bayrammurad Saparov, Tong Zhu, William P. Huhn, Volker Blum, and David B. Mitzi
Chemistry of Materials 2016 Volume 28(Issue 13) pp:4771
Publication Date(Web):June 14, 2016
DOI:10.1021/acs.chemmater.6b01832
Chalcogenides such as CdTe, Cu(In,Ga)(S,Se)2 (CIGSSe), and Cu2ZnSn(S,Se)4 (CZTSSe) have enabled remarkable advances in thin-film photovoltaic performance, but concerns remain regarding (i) the toxicity (CdTe) and (ii) scarcity (CIGSSe/CdTe) of the constituent elements and (iii) the unavoidable antisite disordering that limits further efficiency improvement (CZTSSe). In this work, we show that a different materials class, the BaCu2SnSexS4–x (BCTSSe) system, offers a prospective path to circumvent difficulties (i–iii) and to target new environmentally friendly and earth-abundant absorbers. Antisite disordering and associated band tailing are discouraged in BCTSSe due to the distinct coordination environment of the large Ba2+ cation. Indeed, an abrupt absorption edge and sharp associated photoluminescence emission demonstrate a reduced impact of band tailing in BCTSSe relative to CZTSSe. Our combined experimental and computational studies of BCTSSe reveal that the compositions 0 ≤ x ≤ 4 exhibit a tunable nearly direct or direct bandgap in the 1.6–2 eV range, spanning relevant values for single- or multiple-junction photovoltaic applications. For the first time, a prototype BaCu2SnS4-based thin-film solar cell has been successfully demonstrated, yielding a power conversion efficiency of 1.6% (0.42 cm2 total area). The systematic experimental and theoretical investigations, combined with proof-of-principle device results, suggest promise for BaCu2SnSexS4–x as a thin-film solar cell absorber.
Co-reporter:Donghyeop Shin, Edgard Ngaboyamahina, Yihao Zhou, Jeffrey T. Glass, and David B. Mitzi
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 22) pp:4554-4561
Publication Date(Web):October 28, 2016
DOI:10.1021/acs.jpclett.6b02010
Cu2BaSnS4–xSex films consisting of earth-abundant metals have been examined for photocathode application. Films with different Se contents (i.e., Cu2BaSnS4–xSex with x ≤ 2.4) were synthesized using a cosputter system with post-deposition sulfurization/selenization annealing treatments. Each film adopts a trigonal P31 crystal structure, with progressively larger lattice constants and with band gaps shifting from 2.0 to 1.6 eV, as more Se substitutes for S in the parent compound Cu2BaSnS4. Given the suitable bandgap and earth-abundant elements, the Cu2BaSnS4–xSex films were studied as prospective photocathodes for water splitting. Greater than 6 mA/cm2 was obtained under illumination at −0.4 V versus reversible hydrogen electrode for Pt/Cu2BaSnS4–xSex films with ∼60% Se content (i.e., x = 2.4), whereas a bare Cu2BaSnS4–xSex (x = 2.4) film yielded ∼3 mA/cm2 at −0.4 V/RHE.
Co-reporter:Wiley A. Dunlap-Shohl
The Journal of Physical Chemistry C 2016 Volume 120(Issue 30) pp:16437-16445
Publication Date(Web):July 11, 2016
DOI:10.1021/acs.jpcc.6b05406
Perovskite solar cells with stabilized power conversion efficiency exceeding 15% have been achieved, using a methylammonium lead iodide (MAPbI3) absorber and CdS as the electron transport layer. X-ray photoelectron spectroscopy reveals a small presence of Cd at the surface of most perovskite films fabricated on CdS. Perovskite films were deliberately doped with Cd to understand the possible impacts of Cd diffusion into the perovskite absorber layer. Doping substantially increases the grain size of the perovskite films but also reduces device performance through the formation of an electrical barrier, as inferred by the S-shape of their J–V curves. Time-resolved photoluminescence measurements of the doped films do not indicate substantial nonradiative recombination due to bulk defects, but a secondary phase is evident in these films, which experiments have revealed to be the organic–inorganic hybrid material methylammonium cadmium iodide, (CH3NH3)2CdI4. It is further demonstrated that this compound can form via the reaction of CdS with methylammonium iodide and may form as a competing phase during deposition of the perovskite. Buildup of this insulating compound may act as an electrical barrier at perovskite interfaces, accounting for the drop in device performance.
Co-reporter:Zewen Xiao; Weiwei Meng; Bayrammurad Saparov; Hsin-Sheng Duan; Changlei Wang; Chunbao Feng; Weiqiang Liao; Weijun Ke; Dewei Zhao; Jianbo Wang; David B. Mitzi;Yanfa Yan
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 7) pp:1213-1218
Publication Date(Web):March 14, 2016
DOI:10.1021/acs.jpclett.6b00248
We explore the photovoltaic-relevant properties of the 2D MA2Pb(SCN)2I2 (where MA = CH3NH3+) perovskite using a combination of materials synthesis, characterization and density functional theory calculation, and determine electronic properties of MA2Pb(SCN)2I2 that are significantly different from those previously reported in literature. The layered perovskite with mixed-anions exhibits an indirect bandgap of ∼2.04 eV, with a slightly larger direct bandgap of ∼2.11 eV. The carriers (both electrons and holes) are also found to be confined within the 2D layers. Our results suggest that the 2D MA2Pb(SCN)2I2 perovskite may not be among the most promising absorbers for efficient single-junction solar cell applications; however, use as an absorber for the top cell of a tandem solar cell may still be a possibility if films are grown with the 2D layers aligned perpendicular to the substrates.
Co-reporter:Bayrammurad Saparov, Feng Hong, Jon-Paul Sun, Hsin-Sheng Duan, Weiwei Meng, Samuel Cameron, Ian G. Hill, Yanfa Yan, and David B. Mitzi
Chemistry of Materials 2015 Volume 27(Issue 16) pp:5622
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.chemmater.5b01989
Computational, thin-film deposition, and characterization approaches have been used to examine the ternary halide semiconductor Cs3Sb2I9. Cs3Sb2I9 has two known structural modifications, the 0-D dimer form (space group P63/mmc, no. 194) and the 2-D layered form (P3̅m1, no. 164), which can be prepared via solution and solid-state or gas-phase reactions, respectively. Our computational investigations suggest that the layered form, which is a one-third Sb-deficient derivative of the ubiquitous perovskite structure, is a potential candidate for high-band gap photovoltaic (PV) applications. In this work, we describe details of a two-step deposition approach that enables the preparation of large grain (>1 μm) and continuous thin films of the lead-free layered perovskite derivative Cs3Sb2I9. Depending on the deposition conditions, films that are c-axis oriented or randomly oriented can be obtained. The fabricated thin films show enhanced stability under ambient air, compared to methylammonium lead(II) iodide perovskite films stored under similar conditions, and an optical band gap value of 2.05 eV. Photoelectron spectroscopy study yields an ionization energy of 5.6 eV, with the valence band maximum approximately 0.85 eV below the Fermi level, indicating near-intrinsic, weakly p-type character. Density functional theory (DFT) analysis points to a nearly direct band gap for this material (less than 0.02 eV difference between the direct and indirect band gaps) and a similar high-level of absorption compared to CH3NH3PbI3. The photoluminescence peak intensity of Cs3Sb2I9 is substantially suppressed compared to that of CH3NH3PbI3, likely reflecting the presence of deep level defects that result in nonradiative recombination in the film, with computational results pointing to Ii, ISb, and VI as being likely candidates. A key further finding from this study is that, despite a distinctly layered structure, the electronic transport anisotropy is less pronounced due to the high ionicity of the I atoms and the strong antibonding interactions between the Sb s lone pair states and I p states, which leads to a moderately dispersive valence band.
Co-reporter:Jeehwan Kim;Homare Hiroi;Teodor K. Todorov;Oki Gunawan;Masaru Kuwahara;Tayfun Gokmen;Dhruv Nair;Marinus Hopstaken;Byungha Shin;Yun Seog Lee;Wei Wang;Hiroki Sugimoto
Advanced Materials 2014 Volume 26( Issue 44) pp:7427-7431
Publication Date(Web):
DOI:10.1002/adma.201402373