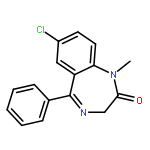
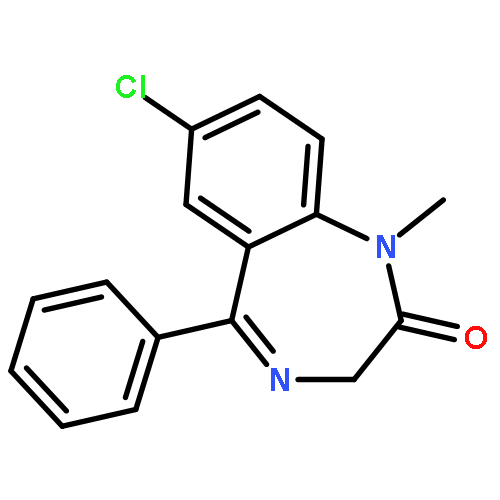
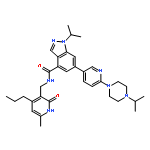
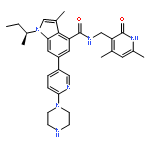
Co-reporter:Anqi Ma ; Wenyu Yu ; Fengling Li ; Rachel M. Bleich ; J. Martin Herold ; Kyle V. Butler ; Jacqueline L. Norris ; Victoria Korboukh ; Ashutosh Tripathy ; William P. Janzen ; Cheryl H. Arrowsmith ; Stephen V. Frye ; Masoud Vedadi ; Peter J. Brown
Journal of Medicinal Chemistry 2014 Volume 57(Issue 15) pp:6822-6833
Publication Date(Web):July 17, 2014
DOI:10.1021/jm500871s
The lysine methyltransferase SETD8 is the only known methyltransferase that catalyzes monomethylation of histone H4 lysine 20 (H4K20). Monomethylation of H4K20 has been implicated in regulating diverse biological processes including the DNA damage response. In addition to H4K20, SETD8 monomethylates non-histone substrates including proliferating cell nuclear antigen (PCNA) and promotes carcinogenesis by deregulating PCNA expression. However, selective inhibitors of SETD8 are scarce. The only known selective inhibitor of SETD8 to date is nahuoic acid A, a marine natural product, which is competitive with the cofactor. Here, we report the discovery of the first substrate-competitive inhibitor of SETD8, UNC0379 (1). This small-molecule inhibitor is active in multiple biochemical assays. Its affinity to SETD8 was confirmed by ITC (isothermal titration calorimetry) and SPR (surface plasmon resonance) studies. Importantly, compound 1 is selective for SETD8 over 15 other methyltransferases. We also describe structure–activity relationships (SAR) of this series.
Co-reporter:Feng Liu ; Fengling Li ; Anqi Ma ; Elena Dobrovetsky ; Aiping Dong ; Cen Gao ; Ilia Korboukh ; Jing Liu ; David Smil ; Peter J. Brown ; Stephen V. Frye ; Cheryl H. Arrowsmith ; Matthieu Schapira ; Masoud Vedadi
Journal of Medicinal Chemistry 2013 Volume 56(Issue 5) pp:2110-2124
Publication Date(Web):February 27, 2013
DOI:10.1021/jm3018332
Protein arginine methyltransferases (PRMTs) play an important role in diverse biological processes. Among the nine known human PRMTs, PRMT3 has been implicated in ribosomal biosynthesis via asymmetric dimethylation of the 40S ribosomal protein S2 and in cancer via interaction with the DAL-1 tumor suppressor protein. However, few selective inhibitors of PRMTs have been discovered. We recently disclosed the first selective PRMT3 inhibitor, which occupies a novel allosteric binding site and is noncompetitive with both the peptide substrate and cofactor. Here we report comprehensive structure–activity relationship studies of this series, which resulted in the discovery of multiple PRMT3 inhibitors with submicromolar potencies. An X-ray crystal structure of compound 14u in complex with PRMT3 confirmed that this inhibitor occupied the same allosteric binding site as our initial lead compound. These studies provide the first experimental evidence that potent and selective inhibitors can be created by exploiting the allosteric binding site of PRMT3.
Co-reporter:Feng Liu ; Dalia Barsyte-Lovejoy ; Fengling Li ; Yan Xiong ; Victoria Korboukh ; Xi-Ping Huang ; Abdellah Allali-Hassani ; William P. Janzen ; Bryan L. Roth ; Stephen V. Frye ; Cheryl H. Arrowsmith ; Peter J. Brown ; Masoud Vedadi
Journal of Medicinal Chemistry 2013 Volume 56(Issue 21) pp:8931-8942
Publication Date(Web):October 8, 2013
DOI:10.1021/jm401480r
Among epigenetic “writers”, “readers”, and “erasers”, the lysine methyltransferases G9a and GLP, which catalyze mono- and dimethylation of histone H3 lysine 9 (H3K9me2) and nonhistone proteins, have been implicated in a variety of human diseases. A “toolkit” of well-characterized chemical probes will allow biological and disease hypotheses concerning these proteins to be tested in cell-based and animal models with high confidence. We previously discovered potent and selective G9a/GLP inhibitors including the cellular chemical probe UNC0638, which displays an excellent separation of functional potency and cell toxicity. However, this inhibitor is not suitable for animal studies due to its poor pharmacokinetic (PK) properties. Here, we report the discovery of the first G9a and GLP in vivo chemical probe UNC0642, which not only maintains high in vitro and cellular potency, low cell toxicity, and excellent selectivity, but also displays improved in vivo PK properties, making it suitable for animal studies.
Co-reporter:Kyle D. Konze, Anqi Ma, Fengling Li, Dalia Barsyte-Lovejoy, Trevor Parton, Christopher J. MacNevin, Feng Liu, Cen Gao, Xi-Ping Huang, Ekaterina Kuznetsova, Marie Rougie, Alice Jiang, Samantha G. Pattenden, Jacqueline L. Norris, Lindsey I. James, Bryan L. Roth, Peter J. Brown, Stephen V. Frye, Cheryl H. Arrowsmith, Klaus M. Hahn, Gang Greg Wang, Masoud Vedadi, and Jian Jin
ACS Chemical Biology 2013 Volume 8(Issue 6) pp:1324
Publication Date(Web):April 8, 2013
DOI:10.1021/cb400133j
EZH2 or EZH1 is the catalytic subunit of the polycomb repressive complex 2 that catalyzes methylation of histone H3 lysine 27 (H3K27). The trimethylation of H3K27 (H3K27me3) is a transcriptionally repressive post-translational modification. Overexpression of EZH2 and hypertrimethylation of H3K27 have been implicated in a number of cancers. Several selective inhibitors of EZH2 have been reported recently. Herein we disclose UNC1999, the first orally bioavailable inhibitor that has high in vitro potency for wild-type and mutant EZH2 as well as EZH1, a closely related H3K27 methyltransferase that shares 96% sequence identity with EZH2 in their respective catalytic domains. UNC1999 was highly selective for EZH2 and EZH1 over a broad range of epigenetic and non-epigenetic targets, competitive with the cofactor SAM and non-competitive with the peptide substrate. This inhibitor potently reduced H3K27me3 levels in cells and selectively killed diffused large B cell lymphoma cell lines harboring the EZH2Y641N mutant. Importantly, UNC1999 was orally bioavailable in mice, making this inhibitor a valuable tool for investigating the role of EZH2 and EZH1 in chronic animal studies. We also designed and synthesized UNC2400, a close analogue of UNC1999 with potency >1,000-fold lower than that of UNC1999 as a negative control for cell-based studies. Finally, we created a biotin-tagged UNC1999 (UNC2399), which enriched EZH2 in pull-down studies, and a UNC1999–dye conjugate (UNC2239) for co-localization studies with EZH2 in live cells. Taken together, these compounds represent a set of useful tools for the biomedical community to investigate the role of EZH2 and EZH1 in health and disease.
Co-reporter:Ilia Korboukh ; Emily A. Hull-Ryde ; Joseph E. Rittiner ; Amarjit S. Randhawa ; Jennifer Coleman ; Brendan J. Fitzpatrick ; Vincent Setola ; William P. Janzen ; Stephen V. Frye ; Mark J. Zylka
Journal of Medicinal Chemistry 2012 Volume 55(Issue 14) pp:6467-6477
Publication Date(Web):June 27, 2012
DOI:10.1021/jm3004834
Adenosine A1 receptor (A1AR) agonists have antinociceptive effects in multiple preclinical models of acute and chronic pain. Although numerous A1AR agonists have been developed, clinical applications of these agents have been hampered by their cardiovascular side effects. Herein we report a series of novel A1AR agonists, some of which are structurally related to adenosine 5′-monophosphate (5′-AMP), a naturally occurring nucleotide that itself activates A1AR. These novel compounds potently activate A1AR in several orthogonal in vitro assays and are subtype selective for A1AR over A2AAR, A2BAR, and A3AR. Among them, UNC32A (3a) is orally active and has dose-dependent antinociceptive effects in wild-type mice. The antinociceptive effects of 3a were completely abolished in A1AR knockout mice, revealing a strict dependence on A1AR for activity. The apparent lack of cardiovascular side effects when administered orally and high affinity (Ki of 36 nM for the human A1AR) make this compound potentially suitable as a therapeutic.
Co-reporter:Xin Chen ; Maria F. Sassano ; Lianyou Zheng ; Vincent Setola ; Meng Chen ; Xu Bai ; Stephen V. Frye ; William C. Wetsel ; Bryan L. Roth
Journal of Medicinal Chemistry 2012 Volume 55(Issue 16) pp:7141-7153
Publication Date(Web):July 30, 2012
DOI:10.1021/jm300603y
Functionally selective G protein-coupled receptor (GPCR) ligands, which differentially modulate canonical and noncanonical signaling, are extremely useful for elucidating key signal transduction pathways essential for both the therapeutic actions and side effects of drugs. However, few such ligands have been created, and very little purposeful attention has been devoted to studying what we term: “structure–functional selectivity relationships” (SFSR). We recently disclosed the first β-arrestin-biased dopamine D2 receptor (D2R) agonists UNC9975 (44) and UNC9994 (36), which have robust in vivo antipsychotic drug-like activities. Here we report the first comprehensive SFSR studies focused on exploring four regions of the aripiprazole scaffold, which resulted in the discovery of these β-arrestin-biased D2R agonists. These studies provide a successful proof-of-concept for how functionally selective ligands can be discovered.
Co-reporter:Feng Liu ; Dalia Barsyte-Lovejoy ; Abdellah Allali-Hassani ; Yunlong He ; J. Martin Herold ; Xin Chen ; Christopher M. Yates ; Stephen V. Frye ; Peter J. Brown ; Jing Huang ; Masoud Vedadi ; Cheryl H. Arrowsmith
Journal of Medicinal Chemistry 2011 Volume 54(Issue 17) pp:6139-6150
Publication Date(Web):July 22, 2011
DOI:10.1021/jm200903z
Protein lysine methyltransferase G9a plays key roles in the transcriptional repression of a variety of genes via dimethylation of lysine 9 on histone H3 (H3K9me2) of chromatin as well as dimethylation of nonhistone proteins including tumor suppressor p53. We previously reported the discovery of UNC0321 (3), the most potent G9a inhibitor to date, via structure-based design and structure–activity relationship (SAR) exploration of the quinazoline scaffold represented by BIX01294 (1). Despite its very high in vitro potency, compound 3 lacks sufficient cellular potency. The design and synthesis of several generations of new analogues aimed at improving cell membrane permeability while maintaining high in vitro potency resulted in the discovery of a number of novel G9a inhibitors such as UNC0646 (6) and UNC0631 (7) with excellent potency in a variety of cell lines and excellent separation of functional potency versus cell toxicity. The design, synthesis, and cellular SAR of these potent G9a inhibitors are described.
Co-reporter:Ángel I. Morales-Ramos, Yue H. Li, Mark Hilfiker, John S. Mecom, Patrick Eidam, Dongchuan Shi, Pei-San Tseng, Carl Brooks, David Zhang, Ning Wang, Jon-Paul Jaworski, Dwight Morrow, Harvey Fries, Richard Edwards, Jian Jin
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 10) pp:2806-2811
Publication Date(Web):15 May 2011
DOI:10.1016/j.bmcl.2011.03.107
Multiple regions of the 3-oxazolidinedione-6-naphthyl-pyridinone series identified via high throughput screening were explored. SAR studies of these regions including the left-hand side oxazolidinedione moiety, α-substituent on the oxazolidinedione ring, central pyridinone core, and substituents on the central pyridinone core led to the discovery of potent EP3 receptor antagonists such as compound 29 which possesses outstanding rat pharmacokinetic properties. Synthesis and SAR of these novel compounds and DMPK properties of representative compounds are discussed.3-Oxazolidinedione-6-napthyl-2-pyridinones are presented as novel and subtype selective EP3 antagonists. The synthesis and extensive SAR that led to the discovery of compound 29 are described.
Co-reporter:Feng Liu ; Xin Chen ; Abdellah Allali-Hassani ; Amy M. Quinn ; Tim J. Wigle ; Gregory A. Wasney ; Aiping Dong ; Guillermo Senisterra ; Irene Chau ; Alena Siarheyeva ; Jacqueline L. Norris ; Dmitri B. Kireev ; Ajit Jadhav ; J. Martin Herold ; William P. Janzen ; Cheryl H. Arrowsmith ; Stephen V. Frye ; Peter J. Brown ; Anton Simeonov ; Masoud Vedadi
Journal of Medicinal Chemistry 2010 Volume 53(Issue 15) pp:5844-5857
Publication Date(Web):July 9, 2010
DOI:10.1021/jm100478y
Protein lysine methyltransferase G9a, which catalyzes methylation of lysine 9 of histone H3 (H3K9) and lysine 373 (K373) of p53, is overexpressed in human cancers. Genetic knockdown of G9a inhibits cancer cell growth, and the dimethylation of p53 K373 results in the inactivation of p53. Initial SAR exploration of the 2,4-diamino-6,7-dimethoxyquinazoline template represented by 3a (BIX01294), a selective small molecule inhibitor of G9a and GLP, led to the discovery of 10 (UNC0224) as a potent G9a inhibitor with excellent selectivity. A high resolution X-ray crystal structure of the G9a−10 complex, the first cocrystal structure of G9a with a small molecule inhibitor, was obtained. On the basis of the structural insights revealed by this cocrystal structure, optimization of the 7-dimethylaminopropoxy side chain of 10 resulted in the discovery of 29 (UNC0321) (Morrison Ki = 63 pM), which is the first G9a inhibitor with picomolar potency and the most potent G9a inhibitor to date.
Co-reporter:Brian Budzik, Vincenzo Garzya, Dongchuan Shi, Graham Walker, Marie Woolley-Roberts, Joanne Pardoe, Adam Lucas, Ben Tehan, Ralph A. Rivero, Christopher J. Langmead, Jeannette Watson, Zining Wu, Ian T. Forbes and Jian Jin
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 6) pp:244
Publication Date(Web):June 8, 2010
DOI:10.1021/ml100105x
Virtual screening of the corporate compound collection yielded compound 1 as a subtype selective muscarinic M1 receptor agonist hit. Initial optimization of the N-capping group of the central piperidine ring resulted in compounds 2 and 3 with significantly improved potency and selectivity. Subsequent optimization of substituents on the phenyl ring of the benzimidazolone moiety led to the discovery of novel muscarinic M1 receptor agonists 4 and 5 with excellent potency, general and subtype selectivity, and pharmacokinetic (PK) properties including good central nervous system (CNS) penetration and oral bioavailability. Compound 5 showed robust in vivo activities in animal models of cognition enhancement. The combination of high potency, excellent selectivity, and good PK properties makes compounds 4 and 5 valuable tool compounds for investigating and validating potential therapeutic benefits resulting from selective M1 activation.Keywords (keywords): 1-(N-substituted piperidin-4-yl)benzimidazolones; benzimidazolones; CNS-penetrant and orally active M1 mAChR agonists; M1 mAChR; M1 muscarinic acetylcholine receptor; subtype selective
Co-reporter:Jian Jin, Ángel Morales-Ramos, Patrick Eidam, John Mecom, Yue Li, Carl Brooks, Mark Hilfiker, David Zhang, Ning Wang, Dongchuan Shi, Pei-San Tseng, Karen Wheless, Brian Budzik, Karen Evans, Jon-Paul Jaworski, Jack Jugus, Lisa Leon, Charlene Wu, Mark Pullen, Bhumika Karamshi, Parvathi Rao, Emma Ward, Nicholas Laping, Christopher Evans, Colin Leach, Dennis Holt, Xin Su, Dwight Morrow, Harvey Fries, Kevin Thorneloe, and Richard Edwards
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 7) pp:316
Publication Date(Web):May 14, 2010
DOI:10.1021/ml100077x
High-throughput screening and subsequent optimization led to the discovery of novel 3-oxazolidinedione-6-aryl-pyridinones exemplified by compound 2 as potent and selective EP3 antagonists with excellent pharmacokinetic properties. Compound 2 was orally active and showed robust in vivo activities in overactive bladder models. To address potential bioactivation liabilities of compound 2, further optimization resulted in compounds 9 and 10, which maintained excellent potency, selectivity, and pharmacokinetic properties and showed no bioactivation liability in glutathione trapping studies. These highly potent, selective, and orally active EP3 antagonists are excellent tool compounds for investigating and validating potential therapeutic benefits from selectively inhibiting the EP3 receptor.Keywords (keywords): 3-oxazolidinedione-6-aryl-pyridinones; EP3 receptor; novel, potent, selective, and orally active antagonists
Co-reporter:Brian Budzik, Vincenzo Garzya, Dongchuan Shi, James J. Foley, Ralph A. Rivero, Christopher J. Langmead, Jeannette Watson, Zining Wu, Ian T. Forbes, Jian Jin
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 12) pp:3540-3544
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmcl.2010.04.128
Biaryl amides were discovered as novel and subtype selective M1 muscarinic acetylcholine receptor agonists. The identification, synthesis, and initial structure–activity relationships that led to compounds 3j and 4c, possessing good M1 agonist potency and intrinsic activity, and subtype selectivity for M1 over M2–5, are described.Biaryl amides were discovered as novel and subtype selective M1 agonists. The identification, synthesis, and initial SAR that led to the discovery of compound 3j are described.
Co-reporter:Brian Budzik, Vincenzo Garzya, Dongchuan Shi, Graham Walker, Yann Lauchart, Adam J. Lucas, Ralph A. Rivero, Christopher J. Langmead, Jeannette Watson, Zining Wu, Ian T. Forbes, Jian Jin
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 12) pp:3545-3549
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmcl.2010.04.127
Further optimization of the biaryl amide series via extensively exploring structure–activity relationships resulted in potent and subtype selective M1 agonists exemplified by compounds 9a and 9j with good rat PK properties including CNS penetration. Synthesis, structure–activity relationships, subtype selectivity for M1 over M2–5, and DMPK properties of these novel compounds are described.Optimization and further SAR exploration of the biaryl amide series resulted in the discovery of orally bioavailable, CNS-penetrant M1 agonists with excellent potency and subtype selectivity.
Co-reporter:Feng Liu ; Xin Chen ; Abdellah Allali-Hassani ; Amy M. Quinn ; Gregory A. Wasney ; Aiping Dong ; Dalia Barsyte ; Ivona Kozieradzki ; Guillermo Senisterra ; Irene Chau ; Alena Siarheyeva ; Dmitri B. Kireev ; Ajit Jadhav ; J. Martin Herold ; Stephen V. Frye ; Cheryl H. Arrowsmith ; Peter J. Brown ; Anton Simeonov ; Masoud Vedadi
Journal of Medicinal Chemistry 2009 Volume 52(Issue 24) pp:7950-7953
Publication Date(Web):November 5, 2009
DOI:10.1021/jm901543m
SAR exploration of the 2,4-diamino-6,7-dimethoxyquinazoline template led to the discovery of 8 (UNC0224) as a potent and selective G9a inhibitor. A high resolution X-ray crystal structure of the G9a−8 complex, the first cocrystal structure of G9a with a small molecule inhibitor, was obtained. The cocrystal structure validated our binding hypothesis and will enable structure-based design of novel inhibitors. 8 is a useful tool for investigating the biology of G9a and its roles in chromatin remodeling.
Co-reporter:Brian Budzik, Yonghui Wang, Dongchuan Shi, Feng Wang, Haibo Xie, Zehong Wan, Chongye Zhu, James J. Foley, Parvathi Nuthulaganti, Lorena A. Kallal, Henry M. Sarau, Dwight M. Morrow, Michael L. Moore, Ralph A. Rivero, Michael Palovich, Michael Salmon, Kristen E. Belmonte, Dramane I. Laine, Jian Jin
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 6) pp:1686-1690
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmcl.2009.01.098
Exploration of multiple regions of a bi-aryl amine template led to the identification of highly potent M3 muscarinic acetylcholine receptor antagonists such as 14 (pA2 = 11.0) possessing good sub-type selectivity for M3 over M2. The structure–activity relationships (SAR) and optimization of the bi-aryl amine series are described.Exploration of multiple regions of a bi-aryl amine template led to the identification of highly potent M3 muscarinic acetylcholine receptor antagonists such as 14 possessing good sub-type selectivity for M3 over M2. The structure–activity relationships and optimization of the bi-aryl amine series are described.
Co-reporter:Jian Jin ; Yonghui Wang ; Dongchuan Shi ; Feng Wang ; Roderick S. Davis ; Qi Jin ; Wei Fu ; James J. Foley ; Edward F. Webb ; Chris J. Dehaas ; Manuela Berlanga ; Miriam Burman ; Henry M. Sarau ; Dwight M. Morrow ; Parvathi Rao ; Lorena A. Kallal ; Michael L. Moore ; Ralph A. Rivero ; Michael Palovich ; Michael Salmon ; Kristen E. Belmonte ;Jakob Busch-Petersen
Journal of Medicinal Chemistry 2008 Volume 51(Issue 16) pp:4866-4869
Publication Date(Web):August 5, 2008
DOI:10.1021/jm800634k
High throughput screening and subsequent optimization led to the discovery of novel quaternary ammonium salts as highly potent muscarinic acetylcholine receptor antagonists with excellent selectivity. Compounds 8a, 13a, and 13b showed excellent inhibitory activity and long duration of action in bronchoconstriction in vivo models in two species via intranasal or intratracheal administration. The novel inhaled muscarinic receptor antagonists are potentially useful therapeutic agents for the treatment of chronic obstructive pulmonary disease and other bronchoconstriction disorders.
Co-reporter:Jian Jin ; Brian Budzik ; Yonghui Wang ; Dongchuan Shi ; Feng Wang ; Haibo Xie ; Zehong Wan ; Chongye Zhu ; James J. Foley ; Edward F. Webb ; Manuela Berlanga ; Miriam Burman ; Henry M. Sarau ; Dwight M. Morrow ; Michael L. Moore ; Ralph A. Rivero ; Michael Palovich ; Michael Salmon ; Kristen E. Belmonte ;Dramane I. Lainé
Journal of Medicinal Chemistry () pp:
Publication Date(Web):September 18, 2008
DOI:10.1021/jm800935u
A series of novel biphenyl piperazines was discovered as highly potent muscarinic acetylcholine receptor antagonists via high throughput screening and subsequent optimization. Compound 5c with respective 500- and 20-fold subtype selectivity for M3 over M2 and M1 exhibited excellent inhibitory activity and long duration of action in a bronchoconstriction in vivo model in mice via intranasal administration. The novel inhaled mAChR antagonists are potentially useful therapeutic agents for the treatment of chronic obstructive pulmonary disease.

![N-[(4,6-dimethyl-2-oxo-1H-pyridin-3-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/1403300/1403254-99-8.png)
![N-[(4,6-dimethyl-2-oxo-1H-pyridin-3-yl)methyl]-3-[ethyl(oxan-4-yl)amino]-2-methyl-5-[4-(morpholin-4-ylmethyl)phenyl]benzamide](http://img.cochemist.com/ccimg/1403300/1403254-99-8_b.png)
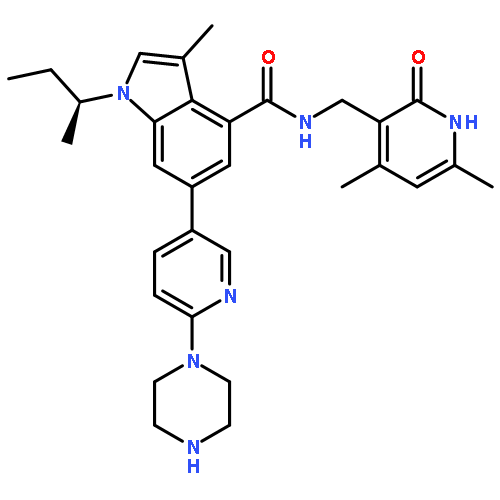
![1-(1-Methylethyl)-N-[(6-methyl-2-oxo-4-propyl-1,2-dihydro-3-pyridinyl)methyl]-6-[2-(4-methyl-1-piperazinyl)-4-pyridinyl]-1H-indazole-4-carboxamide](http://img.cochemist.com/ccimg/1346800/1346704-33-3.png)
![1-(1-Methylethyl)-N-[(6-methyl-2-oxo-4-propyl-1,2-dihydro-3-pyridinyl)methyl]-6-[2-(4-methyl-1-piperazinyl)-4-pyridinyl]-1H-indazole-4-carboxamide](http://img.cochemist.com/ccimg/1346800/1346704-33-3_b.png)
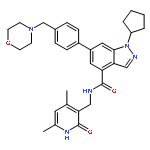
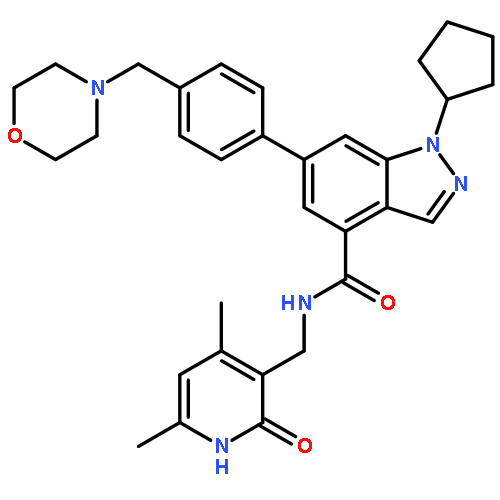








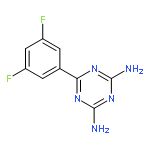
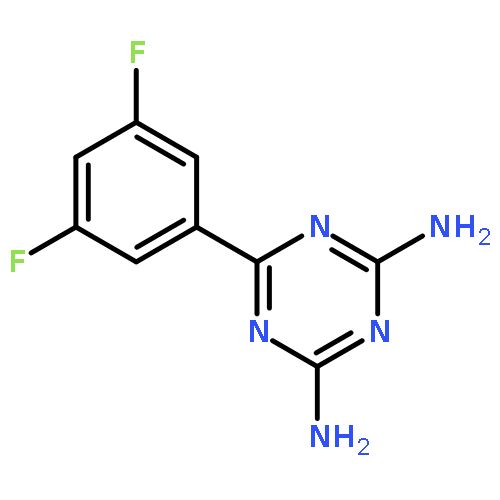
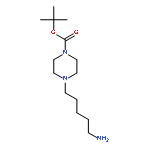
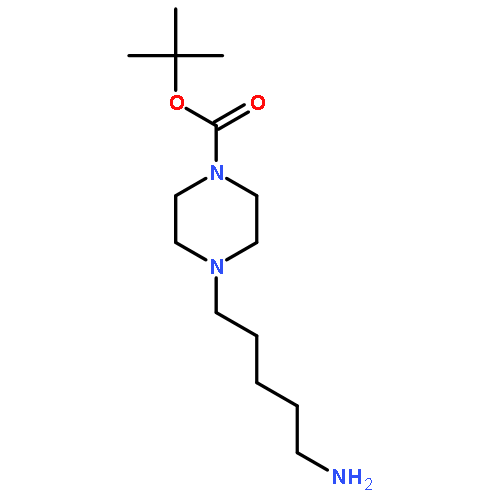
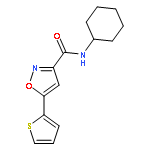
![Benzene, 1-bromo-2-[(1-ethoxyethoxy)methyl]-3,4-difluoro-](http://img.cochemist.com/ccimg/651400/651326-75-9.png)
![Benzene, 1-bromo-2-[(1-ethoxyethoxy)methyl]-3,4-difluoro-](http://img.cochemist.com/ccimg/651400/651326-75-9_b.png)
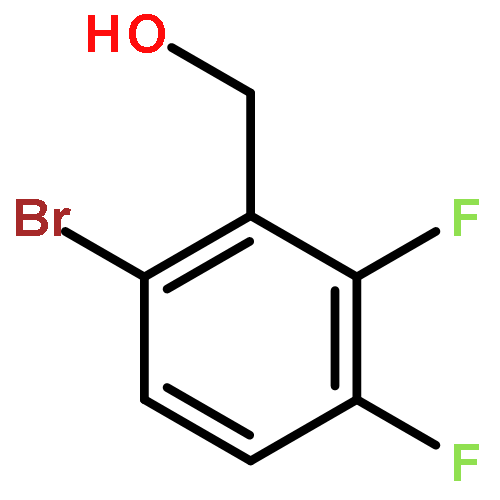
![Benzene, 1-bromo-2-[(1-ethoxyethoxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/651400/651326-66-8.png)
![Benzene, 1-bromo-2-[(1-ethoxyethoxy)methyl]-4-methoxy-](http://img.cochemist.com/ccimg/651400/651326-66-8_b.png)
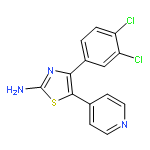
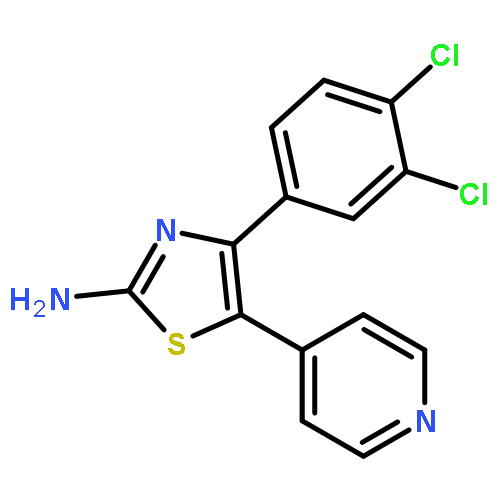
![Cyclohexanol, 4-[(2-phenyl-1H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-,
trans-](http://img.cochemist.com/ccimg/252000/251945-92-3.png)
![Cyclohexanol, 4-[(2-phenyl-1H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-,
trans-](http://img.cochemist.com/ccimg/252000/251945-92-3_b.png)


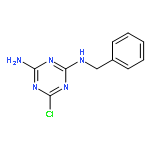








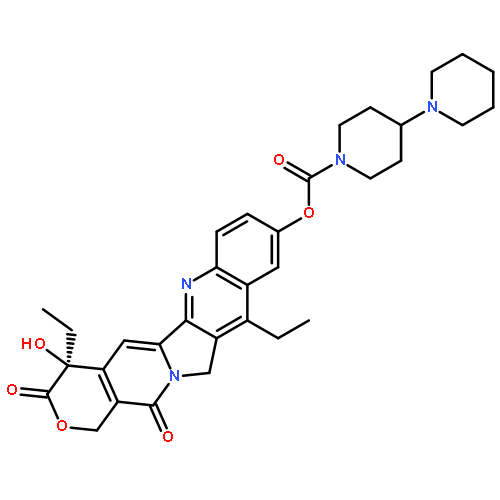
![b-D-Glucopyranosiduronic acid,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-yl](http://img.cochemist.com/ccimg/121100/121080-63-5.png)
![b-D-Glucopyranosiduronic acid,(4S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-yl](http://img.cochemist.com/ccimg/121100/121080-63-5_b.png)




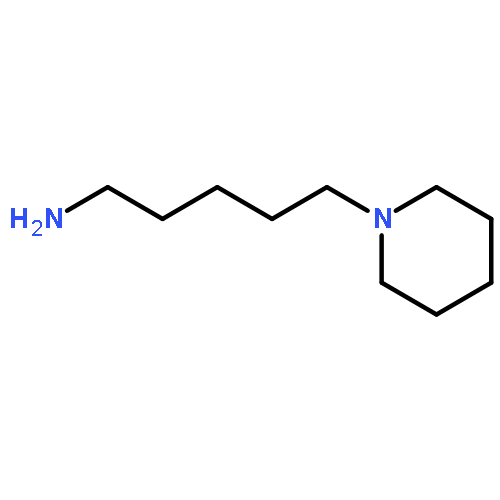
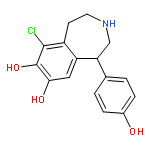
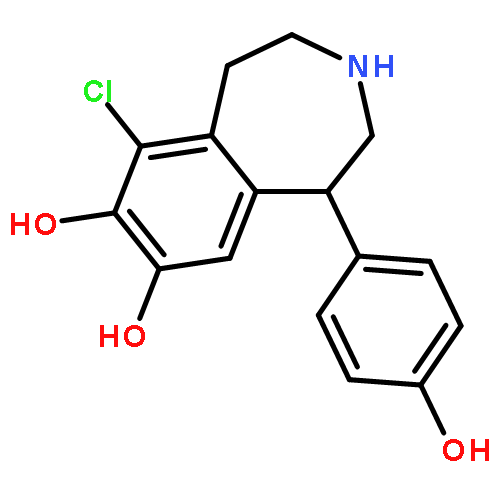
![4-(BUTYLAMINO)-1-ETHYL-6-METHYL 1H-PYRAZOLO[3,4B]PYRIDINE-5-ETHYLCARBOXYLATE HYDROCHLORIDE](http://img.cochemist.com/ccimg/41100/41094-88-6.png)
![4-(BUTYLAMINO)-1-ETHYL-6-METHYL 1H-PYRAZOLO[3,4B]PYRIDINE-5-ETHYLCARBOXYLATE HYDROCHLORIDE](http://img.cochemist.com/ccimg/41100/41094-88-6_b.png)


![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7_b.png)
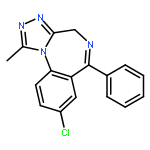









![11H-Dibenz[b,e]azepine,6-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/2000/1977-11-3.png)
![11H-Dibenz[b,e]azepine,6-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/2000/1977-11-3_b.png)