Co-reporter:He Huang, Chenguang Yu, Yueteng Zhang, Yongqiang Zhang, Patrick S. Mariano, and Wei Wang
Journal of the American Chemical Society July 26, 2017 Volume 139(Issue 29) pp:9799-9799
Publication Date(Web):July 10, 2017
DOI:10.1021/jacs.7b05082
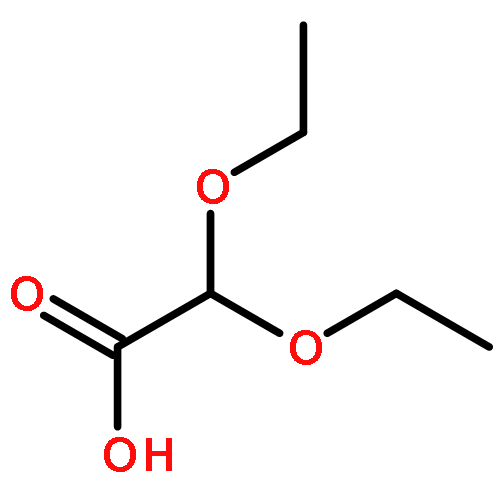
An unprecedented, chemo- and regioselective, organo-photoredox catalyzed hydroformylation reaction of aryl olefins with diethoxyacetic acid as the formylation reagent is described. In contrast to traditional transition metal promoted ionic hydroformylation reactions, the new process follows a unique photoredox promoted, free radical pathway. In this process, a formyl radical equivalent, produced from diethoxacetic acid through a dye (4CzIPN) photocatalyzed, sequential oxidation–decarboxylation route, regio- and chemoselectively adds to a styrene substrate. Importantly, under the optimized reaction conditions the benzylic radical formed in this manner is reduced by SET from the anion radical of 4CzIPN to generate a benzylic anion. Finally, protonation produces the hydroformylation product. By using the new protocol, aldehydes can be generated regioselectively in up to 90% yield. A broad array of functional groups is tolerated in the process, which takes place under mild, metal-free conditions.
Co-reporter:He Huang;Dr. Chenguang Yu;Xiangmin Li;Dr. Yongqiang Zhang;Yueteng Zhang;Dr. Xiaobei Chen; Dr. Patrick S. Mariano;Hexin Xie; Dr. Wei Wang
Angewandte Chemie International Edition 2017 Volume 56(Issue 28) pp:8201-8205
Publication Date(Web):2017/07/03
DOI:10.1002/anie.201703127
AbstractReported herein is a conceptually novel organocatalytic strategy for the formylation of boronic acids. New reactivity is engineered into the α-amino-acid-forming Petasis reaction occurring between aryl boronic acids, amines, and glyoxylic acids to prepare aldehydes. The operational simplicity of the process and its ability to generate structurally diverse and valued aryl, heteroaryl, and α,β-unsaturated aldehydes containing a wide array of functional groups, demonstrates the practical utility of the new synthetic strategy.
Co-reporter:He Huang;Dr. Chenguang Yu;Xiangmin Li;Dr. Yongqiang Zhang;Yueteng Zhang;Dr. Xiaobei Chen; Dr. Patrick S. Mariano;Hexin Xie; Dr. Wei Wang
Angewandte Chemie 2017 Volume 129(Issue 28) pp:8313-8317
Publication Date(Web):2017/07/03
DOI:10.1002/ange.201703127
AbstractReported herein is a conceptually novel organocatalytic strategy for the formylation of boronic acids. New reactivity is engineered into the α-amino-acid-forming Petasis reaction occurring between aryl boronic acids, amines, and glyoxylic acids to prepare aldehydes. The operational simplicity of the process and its ability to generate structurally diverse and valued aryl, heteroaryl, and α,β-unsaturated aldehydes containing a wide array of functional groups, demonstrates the practical utility of the new synthetic strategy.
Co-reporter:Suk Hyun Lim, Dae Won Cho, Jungkweon Choi, Hyunjun An, Jun Ho Shim, Patrick S. Mariano
Tetrahedron 2017 Volume 73, Issue 44(Issue 44) pp:
Publication Date(Web):2 November 2017
DOI:10.1016/j.tet.2017.08.057
Photoaddition reactions of C60 with tertiary N-arylmethyl-N-trimethylsilylmethyl substituted α-aminonitriles were explored. The results show that these photoreactions produce both trimethylsilyl- and cyano group containing fulleropyrrolidines as major products through pathways involving 1,3-dipolar cycloaddition of azomethine ylide intermediates. The ylides are formed either by SET from α-aminonitriles to the triplet excited state of C60 (in N2-purged solutions) followed by desilylation or deprotonation, or by hydrogen atom abstraction by singlet oxygen (in O2-purged solutions). In contrast, photoreactions of C60 with analogous amines that do not contain trimethylsilyl group form fulleropyrrolidines that contain aryl- and cyano substitutents on the pyrrolidine ring. The efficiencies of these photoaddition reactions are influenced by several factors including reaction condition (N2 or O2-purged), solvent polarity, the electronic and structural nature of α-aminonitriles and additive. The presence of trimethylsilyl group in the α-aminonitrile substrates plays a crucial role in enhancing the efficiencies of the fulleropyrrolidine forming reactions.Download high-res image (99KB)Download full-size image
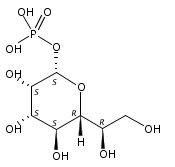
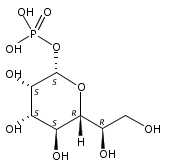
Co-reporter:Chunliang Liu, Debra Dunaway-Mariano, Patrick S. Mariano
European Journal of Medicinal Chemistry 2017 Volume 128(Volume 128) pp:
Publication Date(Web):10 March 2017
DOI:10.1016/j.ejmech.2017.02.001
•A rational design of trehalose 6-phosphate phosphatase (T6PP) inhibitors was tested.•A library of aryl d-glucopyranoside 6-sulfates was synthesized and evaluated.•The inhibition constant of the optimal inhibitor 9a is in the low micromolar range.•The SAR study confirms the aryl d-glucopyranoside 6-sulfate based strategy for the design of T6PP inhibitors.In some organisms, environmental stress triggers trehalose biosynthesis that is catalyzed collectively by trehalose 6-phosphate synthase, and trehalose 6-phosphate phosphatase (T6PP). T6PP catalyzes the hydrolysis of trehalose 6-phosphate (T6P) to trehalose and inorganic phosphate and is a promising target for the development of antibacterial, antifungal and antihelminthic therapeutics. Herein, we report the design, synthesis and evaluation of a library of aryl d-glucopyranoside 6-sulfates to serve as prototypes for small molecule T6PP inhibitors. Steady-state kinetic techniques were used to measure inhibition constants (Ki) of a panel of structurally diverse T6PP orthologs derived from the pathogens Brugia malayi, Ascaris suum, Mycobacterium tuberculosis, Shigella boydii and Salmonella typhimurium. The binding affinities of the most active inhibitor of these T6PP orthologs, 4-n-octylphenyl α-d-glucopyranoside 6-sulfate (9a), were found to be in the low micromolar range. The Ki of 9a with the B. malayi T6PP ortholog is 5.3 ± 0.6 μM, 70-fold smaller than the substrate Michaelis constant. The binding specificity of 9a was demonstrated using several representative sugar phosphate phosphatases from the HAD enzyme superfamily, the T6PP protein fold family of origin. Lastly, correlations drawn between T6PP active site structure, inhibitor structure and inhibitor binding affinity suggest that the aryl d-glucopyranoside 6-sulfate prototypes will find future applications as a platform for development of tailored second-generation T6PP inhibitors.Download high-res image (144KB)Download full-size image
Co-reporter:Suk Hyun Lim, Ho Cheol Jeong, Youngku Sohn, Young-Il Kim, Dae Won Cho, Hee-Jae Woo, Ik-Soo Shin, Ung Chan Yoon, and Patrick S. Mariano
The Journal of Organic Chemistry 2016 Volume 81(Issue 6) pp:2460-2473
Publication Date(Web):February 19, 2016
DOI:10.1021/acs.joc.6b00004
Photoreactions between C60 and secondary N-trimethylsilylmethyl-N-benzylamines were explored to evaluate the feasibility of a new method for secondary aminomethylation of electron acceptors. The results show that photoreactions of C60 with these secondary amines in 10% EtOH-toluene occur to form aminomethyl-1,2-dihydrofullerenes predominantly through a pathway involving single electron transfer (SET)-promoted formation of secondary aminium radicals followed by preferential loss of the α-trimethylsilyl group. The aminomethyl radicals formed in this manner then couple with C60 or C60•– to form radical or anion precursors of the aminomethyl-1,2-dihydrofullerenes. In contrast to thermal and photochemical strategies developed previously, the new SET photochemical approach using α-trimethylsilyl-substituted secondary amines is both mild and efficient, and as a result, it should be useful in broadening the library of substituted fullerenes. Moreover, the results should have an impact on the design of SET-promoted C–C bond forming reactions. Specifically, introduction of an α-trimethylsilyl group leads to a change in the chemoselectivity of SET-promoted reactions of secondary amines with acceptors that typically favor aminium radical N–H deprotonation, leading to N–C bond formation. Finally, symmetric and unsymmetric fulleropyrrolidines are also generated in yields that are highly dependent on the electronic properties of arene ring substituents in amines, irradiation time, and solvent.
Co-reporter:Chunliang Liu, Patrick S. Mariano
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3008-3010
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.10.003
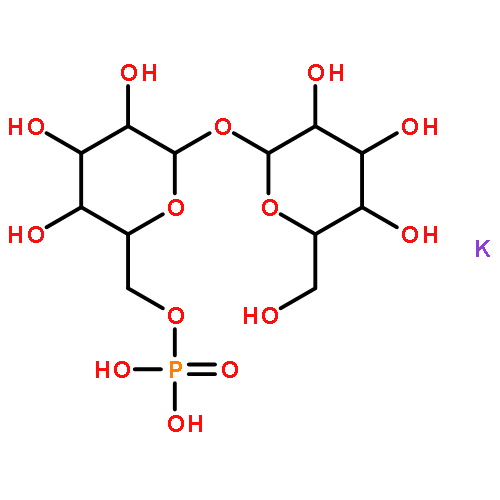
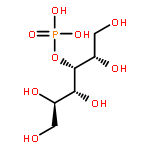
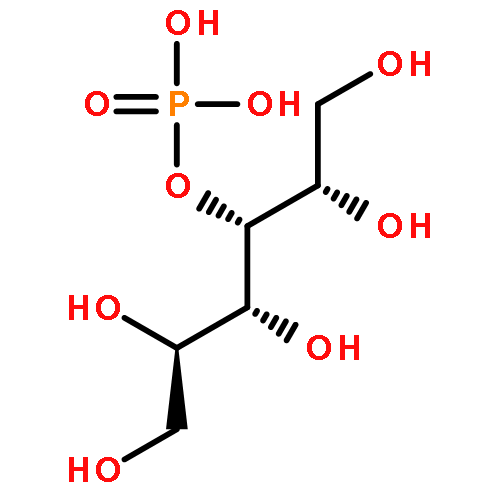
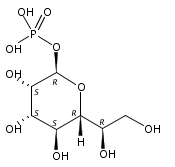
An improved method was developed for the efficient, large-scale synthesis of α,α′-trehalose-6-phosphate starting with α,α′-trehalose. In the sequence, octa-trimethylsilylation of α,α′-trehalose is followed by selective removal of the 6-O-trimethylsilyl protecting group. Reaction of the mono-alcohol with (PhO)2POCl is then followed by simultaneous hydrogenolysis and per-desilylation to generate α,α′-trehalose-6-phosphate, free of contamination by inorganic phosphate.
Co-reporter:Suk Hyun Lim, Jinju Yi, Choon Sup Ra, Keepyung Nahm, Dae Won Cho, Ga Ye Lee, Jinheung Kim, Ung Chan Yoon, Patrick S. Mariano
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3014-3018
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.10.060
Single electron transfer (SET) promoted photoaddition reactions of N-α-trimethylsilylmethyl-N,N-dibenzylamines to fullerene C60 were investigated as part of an effort aimed at developing a general method to prepare various aryl ring containing aminomethylfullerenes and exploring factors that govern photoaddition efficiencies. The results show that the photoaddition reactions take place highly efficiently to form 1,2-adducts. The mechanism for this process involves generation of aminium radicals and C60 anion radical intermediates by a pathway initiated by excited state SET. SET is followed by desilylation of the aminium radicals to produce α-amino radicals that couple with either C60 or its radical anion to form precursors of the 1,2-adducts. The electronic nature of para-substituents on the aryl ring of N-α-trimethylsilylmethyl-N,N-dibenzylamines has a pronounced effect on the efficiency of the photoaddition reaction, with electron donating groups causing a greater than 2-fold enhancement compared to that brought about by electron withdrawing groups.
Co-reporter:Suk Hyun Lim, Woo Sol Lee, Young-Il Kim, Youngku Sohn, Dae Won Cho, Cheolhee Kim, Eunae Kim, John A. Latham, Debra Dunaway-Mariano, Patrick S. Mariano
Tetrahedron 2015 Volume 71(Issue 24) pp:4236-4247
Publication Date(Web):17 June 2015
DOI:10.1016/j.tet.2015.04.077
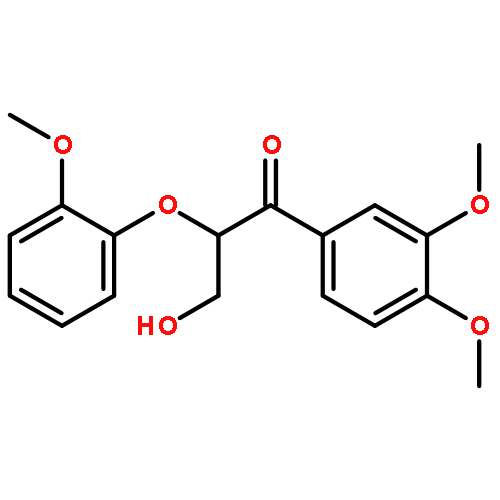
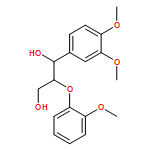
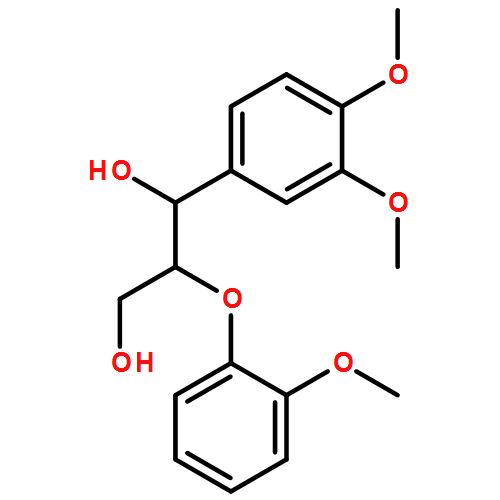
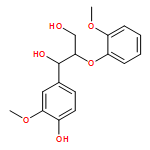
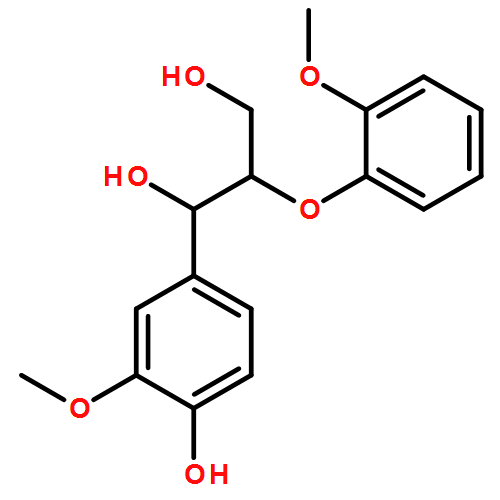
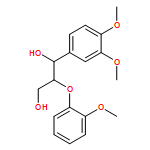
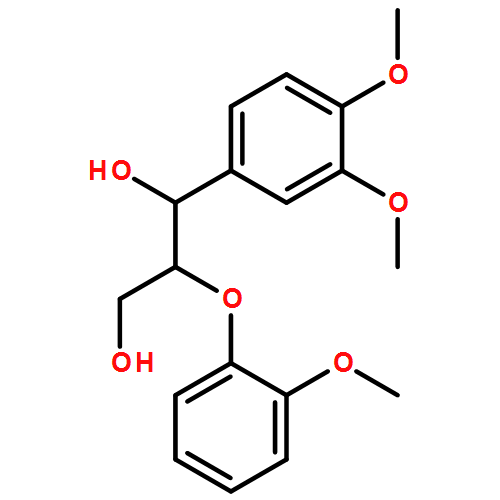
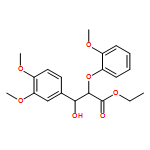
In the current study, 1,2-diarylpropan-1,3-diols, containing varying numbers of methoxy substituents that mimic β-1 type units in lignins, were prepared and subjected to photochemical and enzymatic SET oxidative C–C bond cleavage reactions to explore how product distributions and reactivity profiles depend on the numbers and positions of arene ring methoxy-substituents. For this purpose, product distributions of SET-promoted photochemical reactions of the β-1 model compounds and the characteristics of lignin peroxidase catalyzed bond cleavage reactions of these substances were explored. The results show that both the photochemical and enzymatic reactions, which are known to occur by initial SET to form arylpropanoid cation radicals, generate predominantly aldehydes and β-hydroxyketones through cation radical C1–C2 bond cleavage pathways. In addition, analysis of the relative quantum efficiencies of the SET photochemical processes shows that they do not depend greatly on the numbers and positions of arene ring methoxy substituents of the β-1 model compounds.
Co-reporter:Chun Wu, Debra Dunaway-Mariano, and Patrick S. Mariano
The Journal of Organic Chemistry 2013 Volume 78(Issue 5) pp:1910-1922
Publication Date(Web):October 24, 2012
DOI:10.1021/jo3018473
Pyruvate phosphate dikinase (PPDK) catalyzes the phosphorylation reaction of pyruvate that forms phosphoenolpyruvate (PEP) via two partial reactions: PPDK + ATP + Pi → PPDK-P + AMP + PPi and PPDK-P + pyruvate → PEP + PPDK. Based on its role in the metabolism of microbial human pathogens, PPDK is a potential drug target. A screen of substances that bind to the PPDK ATP-grasp domain active site revealed that flavone analogues are potent inhibitors of the Clostridium symbiosum PPDK. In silico modeling studies suggested that placement of a 3–6 carbon-tethered ammonium substituent at the 3′- or 4′-positions of 5,7-dihydroxyflavones would result in favorable electrostatic interactions with the PPDK Mg-ATP binding site. As a result, polymethylene-tethered amine derivatives of 5,7-dihydroxyflavones were prepared. Steady-state kinetic analysis of these substances demonstrates that the 4′-aminohexyl-5,7-dyhydroxyflavone 10 is a potent competitive PPDK inhibitor (Ki = 1.6 ± 0.1 μM). Single turnover experiments were conducted using 4′-aminopropyl-5,7-dihydroxyflavone 7 to show that this flavone specifically targets the ATP binding site and inhibits catalysis of only the PPDK + ATP + Pi → PPDK-P + AMP PPi partial reaction. Finally, the 4′-aminopbutyl-5,7-dihydroxyflavone 8 displays selectivity for inhibition of PPDK versus other enzymes that utilize ATP and NAD.
Co-reporter:Suk Hyun Lim, Keepyung Nahm, Choon Sup Ra, Dae Won Cho, Ung Chan Yoon, John A. Latham, Debra Dunaway-Mariano, and Patrick S. Mariano
The Journal of Organic Chemistry 2013 Volume 78(Issue 18) pp:9431-9443
Publication Date(Web):August 30, 2013
DOI:10.1021/jo401680z
To gain information about how alkoxy substitution in arene rings of β-O-4 structural units within lignin governs the efficiencies/rates of radical cation C1–C2 bond cleavage reactions, single electron transfer (SET) photochemical and lignin peroxidase-catalyzed oxidation reactions of dimeric/tetrameric model compounds have been explored. The results show that the radical cations derived from less alkoxy-substituted dimeric β-O-4 models undergo more rapid C1–C2 bond cleavage than those of more alkoxy-substituted analogues. These findings gained support from the results of DFT calculations, which demonstrate that C1–C2 bond dissociation energies of β-O-4 radical cations decrease as the degree of alkoxy substitution decreases. In SET reactions of tetrameric compounds consisting of two β-O-4 units, containing different degrees of alkoxy substitution, regioselective radical cation C–C bond cleavage was observed to occur in one case at the C1–C2 bond in the less alkoxy-substituted β-O-4 moiety. However, regioselective C1–C2 cleavage in the more alkoxy-substituted β-O-4 moiety was observed in another case, suggesting that other factors might participate in controlling this process. These observations show that lignins containing greater proportions of less rather than more alkoxylated rings as part of β-O-4 units would be more efficiently cleaved by SET mechanisms.
Co-reporter:Dae Won Cho, Ung Chan Yoon, and Patrick S. Mariano
Accounts of Chemical Research 2011 Volume 44(Issue 3) pp:204
Publication Date(Web):December 27, 2010
DOI:10.1021/ar100125j
Organic photochemists began to recognize in the 1970sthat a new mechanistic pathway involving excited-state single-electron transfer (SET) could be used to drive unique photochemical reactions. Arnold’s seminal studies demonstrated that SET photochemical reactions proceed by way of ion radical intermediates, the properties of which govern the nature of the ensuing reaction pathways. Thus, in contrast to classical photochemical reactions, SET-promoted excited-state processes are controlled by the nature and rates of secondary reactions of intermediate ion radicals. In this Account, we discuss our work in harnessing SET pathways for photochemical synthesis, focusing on the successful production of macrocyclic polyethers, polythioethers, and polyamides.One major thrust of our studies in SET photochemistry has been to develop new, efficient reactions that can be used for the preparation of important natural and non-natural substances. Our efforts with α-silyl donor-tethered phthalimides and naphthalimides have led to the discovery of efficient photochemical processes in which excited-state SET is followed by regioselective formation of carbon-centered radicals. The radical formation takes place through nucleophile-assisted desilylation of intermediate α-silyl-substituted ether-, thioether-, amine-, and amide-centered cation radicals.Early laser flash photolysis studies demonstrated that the rates of methanol- and water-promoted bimolecular desilylations of cation radicals (derived from α-silyl electron donors) exceeded the rates of other cation radical α-fragmentation processes, such as α-deprotonation. In addition, mechanistic analyses of a variety of SET-promoted photocyclization reactions of α-silyl polydonor-linked phthalimides and naphthalimides showed that the chemical and quantum efficiencies of the processes are highly dependent on the lengths and types of the chains connecting the imide acceptor and α-silyl electron donor centers. We also observed that reaction efficiencies are controlled by the rates of desilylation at the α-silyl donor cation radical moieties in intermediate zwitterionic biradicals that are formed by either direct excited-state intramolecular SET or by SET between the donor sites in the intervening chains.It is important to note that knowledge about how these factors govern product yields, regiochemical selectivities, and quantum efficiencies was crucial for the design of synthetically useful photochemical reactions of linked polydonor−acceptor substrates. The fruits of these insights are exemplified by synthetic applications in the concise preparation of cyclic peptide mimics, crown ethers and their lariat- and bis-analogs, and substances that serve as fluorescence sensors for important heavy metal cations.
Co-reporter:Zhimin Li, Zhengang Liu, Dae Won Cho, Jiwen Zou, Maozhen Gong, Robert M. Breece, Andrey Galkin, Ling Li, Hong Zhao, Gabriel D. Maestas, David L. Tierney, Osnat Herzberg, Debra Dunaway-Mariano, Patrick S. Mariano
Journal of Inorganic Biochemistry 2011 Volume 105(Issue 4) pp:509-517
Publication Date(Web):April 2011
DOI:10.1016/j.jinorgbio.2010.12.012
Inhibitors of the Giardia lamblia fructose 1,6-bisphosphate aldolase (GlFBPA), which transforms fructose 1,6-bisphosphate (FBP) to dihydroxyacetone phosphate and glyceraldehyde 3-phosphate, were designed based on 3-hydroxy-2-pyridone and 1,2-dihydroxypyridine scaffolds that position two negatively charged tetrahedral groups for interaction with substrate phosphate binding residues, a hydrogen bond donor to the catalytic Asp83, and a Zn2+ binding group. The inhibition activities for the GlFBPA catalyzed reaction of FBP of the prepared alkyl phosphonate/phosphate substituted 3-hydroxy-2-pyridinones and a dihydroxypyridine were determined. The 3-hydroxy-2-pyridone inhibitor 8 was found to bind to GlFBPA with an affinity (Ki = 14 μM) that is comparable to that of FBP (Km = 2 μM) or its inert analog TBP (Ki = 1 μM). The X-ray structure of the GlFBPA–inhibitor 8 complex (2.3 Å) shows that 8 binds to the active site in the manner predicted by in silico docking with the exception of coordination with Zn2+. The observed distances and orientation of the pyridone ring O=C–C–OH relative to Zn2+ are not consistent with a strong interaction. To determine if Zn2+coordination occurs in the GlFBPA–inhibitor 8 complex in solution, EXAFS spectra were measured. A four coordinate geometry comprised of the three enzyme histidine ligands and an oxygen atom from the pyridone ring O=C–C–OH was indicated. Analysis of the Zn2+ coordination geometries in recently reported structures of class II FBPAs suggests that strong Zn2+ coordination is reserved for the enediolate-like transition state, accounting for minimal contribution of Zn2+ coordination to binding of 8 to GlFBPA.Stereoviews of the active site of the complex between 3-hydroxy-2-pyridone inhibitor 8 and GlFBPA as determined by X-ray crystallography.
Co-reporter:Dae Won Cho, John A. Latham, Hea Jung Park, Ung Chan Yoon, Paul Langan, Debra Dunaway-Mariano, and Patrick S. Mariano
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2840-2852
Publication Date(Web):March 8, 2011
DOI:10.1021/jo200253v
New types of tetrameric lignin model compounds, which contain the common β-O-4 and β-1 structural subunits found in natural lignins, have been prepared and carbon−carbon bond fragmentation reactions of their cation radicals, formed by photochemical (9,10-dicyanoanthracene) and enzymatic (lignin peroxidase) SET-promoted methods, have been explored. The results show that cation radical intermediates generated from the tetrameric model compounds undergo highly regioselective C−C bond cleavage in their β-1 subunits. The outcomes of these processes suggest that, independent of positive charge and odd-electron distributions, cation radicals of lignins formed by SET to excited states of sensitizers or heme-iron centers in enzymes degrade selectively through bond cleavage reactions in β-1 vs β-O-4 moieties. In addition, the findings made in the enzymatic studies demonstrate that the sterically large tetrameric lignin model compounds undergo lignin peroxidase-catalyzed cleavage via a mechanism involving preliminary formation of an enzyme−substrate complex.
Co-reporter:Liangbing Wang, Hua Huang, Henry H. Nguyen, Karen N. Allen, Patrick S. Mariano and Debra Dunaway-Mariano
Biochemistry 2010 Volume 49(Issue 6) pp:
Publication Date(Web):January 5, 2010
DOI:10.1021/bi902018y
d-glycero-d-manno-Heptose-1,7-bisphosphate phosphatase (GmhB) is a member of the histidinol-phosphate phosphatase (HisB) subfamily of the haloalkanoic acid dehalogenase (HAD) enzyme superfamily. GmhB supports two divergent biochemical pathways in bacteria: the d-glycero-d-manno-heptose-1α-GDP pathway (in S-layer glycoprotein biosynthesis) and the l-glycero-d-manno-heptose-1β-ADP pathway (in lipid A biosynthesis). Herein, we report the comparative analysis of substrate recognition in selected GmhB orthologs. The substrate specificity of the l-glycero-d-manno-heptose-1β-ADP pathway GmhB from Escherichia coli K-12 was evaluated using hexose and heptose bisphosphates, histidinol phosphate, and common organophosphate metabolites. Only d-glycero-d-manno-heptose 1β,7-bisphosphate (kcat/Km = 7 × 106 M−1 s−1) and d-glycero-d-manno-heptose 1α,7-bisphosphate (kcat/Km = 7 × 104 M−1 s−1) displayed physiologically significant substrate activity. 31P NMR analysis demonstrated that E. coli GmhB selectively removes the C(7) phosphate. Steady-state kinetic inhibition studies showed that d-glycero-d-manno-heptose 1β-phosphate (Kis = 60 μM, and Kii = 150 μM) and histidinol phosphate (Kis = 1 mM, and Kii = 6 mM), while not hydrolyzed, do in fact bind to E. coli GmhB, which leads to the conclusion that nonproductive binding contributes to substrate discrimination. High catalytic efficiency and a narrow substrate range are characteristic of a well-evolved metabolic enzyme, and as such, E. coli GmhB is set apart from most HAD phosphatases (which are typically inefficient and promiscuous). The specialization of the biochemical function of GmhB was examined by measuring the kinetic constants for hydrolysis of the α- and β-anomers of d-glycero-d-manno-heptose 1β,7-bisphosphate catalyzed by the GmhB orthologs of the l-glycero-d-manno-heptose 1β-ADP pathways operative in Bordetella bronchiseptica and Mesorhizobium loti and by the GmhB of the d-glycero-d-manno-heptose 1α-GDP pathway operative in Bacteroides thetaiotaomicron. The results show that although each of these representatives possesses physiologically significant catalytic activity toward both anomers, each displays substantial anomeric specificity. Like E. coli GmhB, B. bronchiseptica GmhB and M. loti GmhB prefer the β-anomer, whereas B. thetaiotaomicron GmhB is selective for the α-anomer. By determining the anomeric configuration of the physiological substrate (d-glycero-d-manno-heptose 1,7-bisphosphate) for each of the four GmhB orthologs, we discovered that the anomeric specificity of GmhB correlates with that of the pathway kinase. The conclusion drawn from this finding is that the evolution of the ancestor to GmhB in the HisB subfamily provided for specialization toward two distinct biochemical functions.
Co-reporter:Jiwen Zou, Dae Won Cho, Patrick S. Mariano
Tetrahedron 2010 66(32) pp: 5955-5961
Publication Date(Web):
DOI:10.1016/j.tet.2010.06.027
Co-reporter:Jiwen Zou and Patrick S. Mariano
Photochemical & Photobiological Sciences 2008 vol. 7(Issue 4) pp:393-404
Publication Date(Web):03 Mar 2008
DOI:10.1039/B801808C
The discovery in the 1970s by Kaplan, Wilzbach and Pavlik that pyridinium salts undergo a unique cyclization reaction to produce bicyclic-aziridines was virtually unrecognized for nearly three decades. It was only comparatively recently that the process was explored in more detail and its synthetic potential exploited. In this Perspective, photocyclization reactions of pyridinium salts will be discussed, starting with the initial discovery, covering related processes of pyrylium salts, and extending to applications to the synthesis of natural and non-natural products of biomedical interest.
Co-reporter:Runtang Wang, Zhiming Zhao, Patrick S. Mariano, Kyung Hwa Choi, Sang Ha Kim, Ung Chan Yoon
Journal of Photochemistry and Photobiology A: Chemistry 2005 Volume 175(2–3) pp:232-241
Publication Date(Web):31 October 2005
DOI:10.1016/j.jphotochem.2005.05.005
A novel method has been developed for the synthesis of naphthalene chromophore containing, lariat-type crown ethers. The route employs SET-promoted photocyclization reactions of polyether-tethered 2,3-naphthalimides to generate the variously ring-sized crown ether cores and an allylsilane N-acyliminum ion addition process to install amino ether side chains. The metal cation binding properties of the lariat-crown ethers, prepared in this manner, were evaluated. In addition, the ability of the lariat-crown ethers to serve as SET-based, fluorescence sensors of metal cations was probed. The results show that although the novel lariat-crown ethers strongly complex alkali metal cations (Na, K, Rb, Cs), this complexation is not associated with enhanced fluorescence from the naphthalene chromophores as would be expected if cation binding impeded SET quenching by the tertiary amine donor in the side chains. In contrast, the novel lariat-crown ethers serve as sensitive sensors for the divalent metal cations of Mg and Cu and the monovalent cation of Ag.
Co-reporter:Chunliang Liu, Debra Dunaway-Mariano, Patrick S. Mariano
Tetrahedron (9 March 2017) Volume 73(Issue 10) pp:1324-1330
Publication Date(Web):9 March 2017
DOI:10.1016/j.tet.2017.01.041
Co-reporter:Ho Cheol Jeong, Suk Hyun Lim, Youngku Sohn, Young-Il Kim, Hoeun Jang, Dae Won Cho, Patrick S. Mariano
Tetrahedron Letters (8 March 2017) Volume 58(Issue 10) pp:
Publication Date(Web):8 March 2017
DOI:10.1016/j.tetlet.2017.01.073
•SET-promoted photoaddition reaction between C60 and benzylamine derivatives were explored.•Electronic and steric effects of benzylamines influence efficiencies of photoaddition reaction.•Reaction efficiencies are largely governed by electronic effects of benzylamines.•Steric effects play a significant role in electron deficient benzylamine derivatives.Single electron transfer (SET)-promoted photoaddition reactions between fullerene C60 and both various alkyl (Me, Et, i-Pr, t-Bu)- and para-substituted (p-Me, p-OMe, p-F, p-CF3) arene ring containing, N-α-trimethylsilyl-N-alkyl-N-benzylamines were explored to gain information about photoproduct profiles and how the electronic and steric nature of the amine substrates influence reaction efficiencies. The results show that visible light (λ > 540 nm) irradiation of 10% EtOH-toluene solutions containing C60 and N-α-trimethylsilyl-N-alkyl-N-benzylamines produce 1-aminomethyl-1,2-dihydrofullerenes as a sole photoproduct. In addition, SET-promoted photoaddition reactions of unsubstituted and para-electron donating group substituted arene ring containing N-α-trimethylsilyl-N-alkyl-N-benzylamines take place to give photoproducts more efficiently than those containing para-electron withdrawing group substituted arene rings. Moreover, although steric factors are less significant than the electronic nature of the amine substrates in governing reaction efficiencies, sterics do play a significant role in photoreactions of electron deficient amine substrates.
Co-reporter:Ho Cheol Jeong, Suk Hyun Lim, Dae Won Cho, Sung Hong Kim and Patrick S. Mariano
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 44) pp:NaN10510-10510
Publication Date(Web):2016/10/18
DOI:10.1039/C6OB02069K
Single electron transfer (SET) promoted photoaddition reactions of secondary N-α-trimethylsilyl-N-alkylamines to C60 were explored to gain a deeper understanding of the mechanistic pathways followed and to expand the library of novel types of organofullerenes that can be generated using this approach. The results show that photoreactions of 10% EtOH–toluene solutions containing C60 and N-α-trimethylsilyl-N-alkylamines produce either aminomethyl-1,2-dihydrofullerenes or symmetric fulleropyrrolidines as major products depending on the nature of alkyl substituents. In contrast, photoreactions of 10% EtOH–ODCB solutions of these amines with C60 mainly lead to the formation of symmetric fulleropyrrolidines. Based on the analysis of product distributions and the results of earlier studies, two feasible mechanistic pathways are proposed for these processes. One route is initiated by SET from the amine substrates to the triplet-excited state of C60 to form the corresponding aminium radicals and C60 anion radicals. EtOH-promoted desilylation of the aminium radicals then takes place to produce aminomethyl radicals which can either add to C60 or couple with the C60 radical anions to form respective radicals or anion precursors of aminomethyl-1,2-dihydrofullerene products. The competing pathway leading to the generation of symmetric fulleropyrrolidines also involves the formation of aminomethyl radicals by using the sequential SET-desilylation process. In this route, the aminomethyl radicals are oxidized by SET to C60 to form iminium ions, which are then transformed to azomethine ylides by a pathway involving a second molecule of the secondary amine. Dipolar cycloaddition of the azomethine ylides to C60 forms the symmetric fulleropyrrolidine cycloadducts. Importantly, the observation that symmetric fulleropyrrolidines are the sole products formed in photoreactions between N-α-trimethylsilyl-N-alkylamines and C60 in 10% EtOH–ODCB has synthetic significance.
.jpg)





![3-METHYL-5,6,7,8-TETRAHYDROIMIDAZO[1,5-A]PYRAZINE](http://img.cochemist.com/ccimg/333400/333333-34-9.png)
![3-METHYL-5,6,7,8-TETRAHYDROIMIDAZO[1,5-A]PYRAZINE](http://img.cochemist.com/ccimg/333400/333333-34-9_b.png)






![2-Propenal, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/95200/95123-61-8.png)
![2-Propenal, 3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/95200/95123-61-8_b.png)
![Estra-1,3,5(10)-trien-17-one, 3-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/92900/92817-04-4.png)
![Estra-1,3,5(10)-trien-17-one, 3-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/92900/92817-04-4_b.png)
![(1r)-1-[(3ar,4s,6r)-2,2-dimethyl-4-phenylmethoxy-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxol-6-yl]-2-trityloxyethanol](http://img.cochemist.com/ccimg/91400/91364-11-3.png)
![(1r)-1-[(3ar,4s,6r)-2,2-dimethyl-4-phenylmethoxy-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxol-6-yl]-2-trityloxyethanol](http://img.cochemist.com/ccimg/91400/91364-11-3_b.png)






![Benzene, 1-[(1E)-2-bromoethenyl]-4-methoxy-](http://img.cochemist.com/ccimg/27600/27570-08-7.png)
![Benzene, 1-[(1E)-2-bromoethenyl]-4-methoxy-](http://img.cochemist.com/ccimg/27600/27570-08-7_b.png)






![Benzoic acid, 4-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/17800/17763-71-2.png)
![Benzoic acid, 4-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](http://img.cochemist.com/ccimg/17800/17763-71-2_b.png)
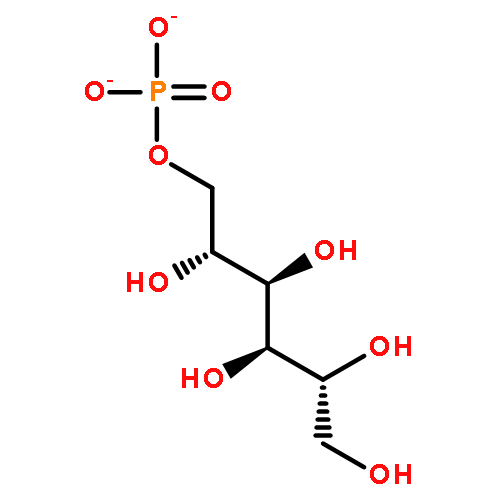
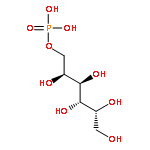



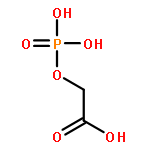

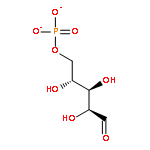




















![Benzene, [(1Z)-2-bromoethenyl]-](/data/chemimg/1121700/588-73-8.png)
![Benzene, [(1Z)-2-bromoethenyl]-](/data/chemimg/1121700/588-73-8_b.png)
![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7.png)
![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7_b.png)






















![8-(1H-IMIDAZOL-4-YLMETHYLENE)-6,8-DIHYDRO-THIAZOLO[5,4-E]INDOL-7-ONE](http://img.cochemist.com/ccimg/608600/608512-97-6.png)
![8-(1H-IMIDAZOL-4-YLMETHYLENE)-6,8-DIHYDRO-THIAZOLO[5,4-E]INDOL-7-ONE](http://img.cochemist.com/ccimg/608600/608512-97-6_b.png)













![1H-Indole-5-sulfonamide, 3-[2-[4-(aminosulfonyl)phenyl]hydrazinylidene]-2,3-dihydro-2-oxo-](/data/chemimg/124900/222035-15-6.png)
![1H-Indole-5-sulfonamide, 3-[2-[4-(aminosulfonyl)phenyl]hydrazinylidene]-2,3-dihydro-2-oxo-](/data/chemimg/124900/222035-15-6_b.png)










![Benzenesulfonamide,N-[2-[[[3-(4-chlorophenyl)-2-propen-1-yl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxy-](http://img.cochemist.com/ccimg/139300/139298-40-1.png)
![Benzenesulfonamide,N-[2-[[[3-(4-chlorophenyl)-2-propen-1-yl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-methoxy-](http://img.cochemist.com/ccimg/139300/139298-40-1_b.png)
![Methanesulfonic acid, trifluoro-, [bis(phenylmethoxy)phosphinyl]methylester](http://img.cochemist.com/ccimg/131300/131292-83-6.png)
![Methanesulfonic acid, trifluoro-, [bis(phenylmethoxy)phosphinyl]methylester](http://img.cochemist.com/ccimg/131300/131292-83-6_b.png)
![Ethanol,2-[[9-methyl-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/101700/101622-51-9.png)
![Ethanol,2-[[9-methyl-6-[(phenylmethyl)amino]-9H-purin-2-yl]amino]-](http://img.cochemist.com/ccimg/101700/101622-51-9_b.png)
![9,12-Epoxy-1H-diindolo[1,2,3-fg:3',2',1'-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid, 2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, methyl ester,(9S,10R,12R)-](http://img.cochemist.com/ccimg/99600/99533-80-9.png)
![9,12-Epoxy-1H-diindolo[1,2,3-fg:3',2',1'-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid, 2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, methyl ester,(9S,10R,12R)-](http://img.cochemist.com/ccimg/99600/99533-80-9_b.png)







![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)




























![Thiazolium,3-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-4-methyl-5-(4,6,6-trihydroxy-4,6-dioxido-3,5-dioxa-4,6-diphosphahex-1-yl)-,inner salt (9CI)](http://img.cochemist.com/ccimg/200/136-09-4.png)
![Thiazolium,3-[(4-amino-2-methyl-5-pyrimidinyl)methyl]-4-methyl-5-(4,6,6-trihydroxy-4,6-dioxido-3,5-dioxa-4,6-diphosphahex-1-yl)-,inner salt (9CI)](http://img.cochemist.com/ccimg/200/136-09-4_b.png)