Co-reporter:Zhao Dang, Hua Xie, Lei Zhu, Qingye Zhang, Zhijun Li, Li Huang, and Chin-Ho Chen
ACS Medicinal Chemistry Letters November 9, 2017 Volume 8(Issue 11) pp:1199-1199
Publication Date(Web):October 9, 2017
DOI:10.1021/acsmedchemlett.7b00376
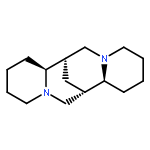
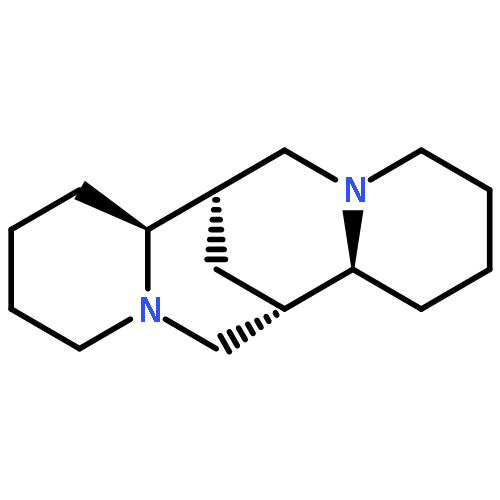
As a step toward developing novel anti-HIV agents, we have identified a class of quinolizidines, including aloperine, that inhibit HIV at 1–5 μM by blocking viral entry. In this study, we have optimized the structure of aloperine and derived compounds with markedly improved activity. Our structural optimization has yielded an aloperine derivative 19 with approximately a 15-fold increase in anti-HIV-1 activity. Our mechanism of action study reveals that compound 19 does not inhibit binding of HIV-1 to receptors but arrests the virus from fusion with the membrane. Binding of the compound to HIV-1gp120 might be responsible for its anti-HIV-1 entry activity.Keywords: aloperine; entry inhibitors; HIV entry;
Co-reporter:Zhao Dang, Lei Zhu, Weihong Lai, Hal Bogerd, Kuo-Hsiung Lee, Li Huang, and Chin-Ho Chen
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 3) pp:240
Publication Date(Web):January 9, 2016
DOI:10.1021/acsmedchemlett.5b00339
A quinolizidine-type alkaloid aloperine was found to inhibit HIV-1 infection by blocking HIV-1 entry. Aloperine inhibited HIV-1 envelope-mediated cell–cell fusion at low micromolar concentrations. To further improve the antiviral potency, more than 30 aloperine derivatives with a variety of N12-substitutions were synthesized. Among them, 12d with an N-(1-butyl)-4-trifluoromethoxy-benzamide side chain showed the most potent anti-HIV-1 activity with EC50 at 0.69 μM. Aloperine derivatives inhibited both X4 and R5 HIV-1 Env-mediated cell–cell fusions. In addition, both BMS-806, a compound representing a class of HIV-1 gp120-targeting small molecules in clinical trials, and resistant and sensitive HIV-1 Env-mediated cell–cell fusions were equally sensitive to aloperine derivatives. These results suggest that aloperine and its derivatives are a new class of anti-HIV-1 entry inhibitors.Keywords: aloperine; entry inhibitor; HIV-1
Co-reporter:Weihong Lai; Li Huang; Lei Zhu; Guido Ferrari; Cliburn Chan; Wei Li; Kuo-Hsiung Lee
Journal of Medicinal Chemistry 2015 Volume 58(Issue 21) pp:8638-8646
Publication Date(Web):October 28, 2015
DOI:10.1021/acs.jmedchem.5b01233
HIV-1-latency-reversing agents, such as histone deacetylase inhibitors (HDACIs), were ineffective in reducing latent HIV-1 reservoirs ex vivo using CD4 cells from patients as a model. This deficiency poses a challenge to current pharmacological approaches for HIV-1 eradication. The results of this study indicated that gnidimacrin (GM) was able to markedly reduce the latent HIV-1 DNA level and the frequency of latently infected cells in an ex vivo model using patients peripheral blood mononuclear cells. GM induced approximately 10-fold more HIV-1 production than the HDACI SAHA or romidepsin, which may be responsible for the effectiveness of GM in reducing latent HIV-1 levels. GM achieved these effects at low picomolar concentrations by selective activation of protein kinase C βI and βII. Notably, GM was able to reduce the frequency of HIV-1 latently infected cells at concentrations without global T cell activation or stimulating inflammatory cytokine production. GM merits further development as a clinical trial candidate for latent HIV-1 eradication.
Co-reporter:Zhao Dang, Katherine Jung, Lei Zhu, Hua Xie, Kuo-Hsiung Lee, Chin-Ho Chen, and Li Huang
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 3) pp:355
Publication Date(Web):January 29, 2015
DOI:10.1021/ml500533x
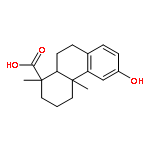
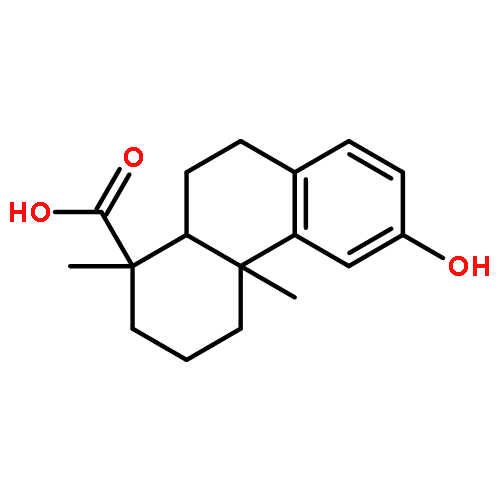
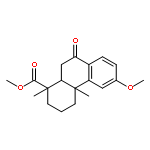
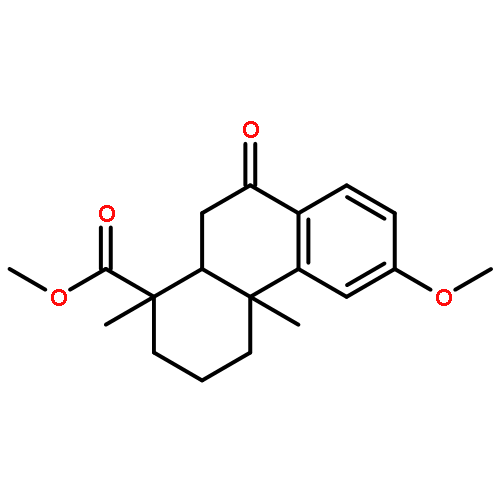
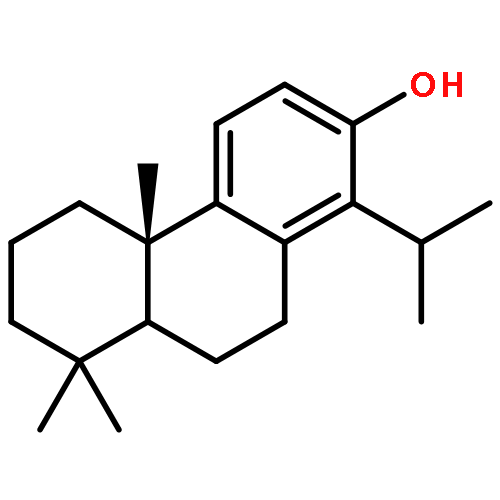
A series of diterpenoid derivatives based on podocarpic acid were synthesized and evaluated as anti-influenza A virus agents. Several of the novel podocarpic acid derivatives exhibited nanomolar activities against an H1N1 influenza A virus (A/Puerto Rico/8/34) that was resistant to two anti-influenza drugs, oseltamivir and amantadine. This class of compounds inhibits the influenza virus by targeting the viral hemagglutinin-mediated membrane fusion. These results indicated that podocarpic acid derivatives may serve as potential drug candidates to fight drug-resistant influenza A virus infections.Keywords: hemagglutinin; Influenza A; influenza inhibitor; podocarpic acid; totarol
Co-reporter:Zhao Dang, Katherine Jung, Lei Zhu, Weihong Lai, Hua Xie, Kuo-Hsiung Lee, Li Huang, and Chin-Ho Chen
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 8) pp:942
Publication Date(Web):June 30, 2014
DOI:10.1021/ml500236n
Influenza A virus infection causes a contagious respiratory illness that poses a threat to human health. However, there are limited anti-influenza A therapeutics available, which is further compounded by the emergence of drug resistant viruses. In this study, Sophora quinolizidine alkaloids were identified as a new class of anti-influenza A virus agents. Among the tested Sophora alkaloids, dihydroaloperine exhibited the most potent activity with an EC50 of 11.2 μM. The potency of the quinolizidine alkaloids was improved by approximately 5-fold with chemical modifications on the aloperine molecule. These compounds were effective against an H1N1 influenza A virus that was resistant to the two antiflu drugs oseltamivir and amantadine. The identification of the quinolizidine alkaloids as effective and novel anti-influenza A agents may aid in the development of new therapeutics.Keywords: Influenza; influenza inhibitor; nucleoprotein
Co-reporter:Zhao Dang ; Phong Ho ; Lei Zhu ; Keduo Qian ; Kuo-Hsiung Lee ; Li Huang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 5) pp:2029-2037
Publication Date(Web):February 4, 2013
DOI:10.1021/jm3016969
Bevirimat (1, BVM) is an anti-HIV agent that blocks HIV-1 replication by interfering with HIV-1 Gag-SP1 processing at a late stage of viral maturation. However, clinical trials of 1 have revealed a high baseline drug resistance that is attributed to naturally occurring polymorphisms in HIV-1 Gag. To overcome the drug resistance, 28 new derivatives of 1 were synthesized and tested against compound 1-resistant (BVM-R) HIV-1 variants. Among them, compound 6 exhibited much improved activity against several HIV-1 strains carrying BVM-R polymorphisms. Compound 6 was at least 20-fold more potent than 1 against the replication of NL4-3/V370A, which carries the most prevalent clinical BVM-R polymorphism in HIV-1 Gag-SP1. Thus, compound 6 merits further development as a potential anti-AIDS clinical trial candidate.
Co-reporter:Zhao Dang, Kathy Jung, Keduo Qian, Kuo-Hsiung Lee, Li Huang, and Chin-Ho Chen
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 11) pp:925
Publication Date(Web):August 15, 2012
DOI:10.1021/ml3001962
Accumulation of aberrant protein aggregates, such as amyloid β peptide (Aβ), due to decreased proteasome activities, might contribute to the neurodegeneration in Alzheimer's disease. In this study, lithocholic acid derivatives 3α-O-pimeloyl-lithocholic acid methyl ester (2) and its isosteric isomer (6) were found to activate the chymotrypsin-like activity of the proteasome at an EC50 of 7.8 and 4.3 μM, respectively. Replacing the C24 methyl ester in 2 with methylamide resulted in a complete devoid of proteasome activating activity. Epimerizing the C3 substituent from an α to β orientation transformed the activator into a proteasome inhibitor. Unlike the cellular proteasome activator PA28, proteasome activated by 2 was not inhibited by Aβ. Furthermore, 2 potently antagonized the inhibitory effect of Aβ on the proteasome. In summary, compound 2 represents a novel class of small molecules that not only activates the proteasome but also antagonizes the inhibitory effect of Aβ on the proteasome.Keywords: Alzheimer's disease; amyloid β; lithocholic acid; proteasome activator
Co-reporter:Zhao Dang, Keduo Qian, Phong Ho, Lei Zhu, Kuo-Hsiung Lee, Li Huang, Chin-Ho Chen
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 16) pp:5190-5194
Publication Date(Web):15 August 2012
DOI:10.1016/j.bmcl.2012.06.080
Betulinic acid derivatives modified at the C28 position are HIV-1entry inhibitors such as compound A43D; however, modified at the C3 position instead of C28 give HIV-1 maturation inhibitor such as bevirimat. Bevirimat exhibited promising pharmacokinetic profiles in clinical trials, but its effectiveness was compromised by the high baseline drug resistance of HIV-1 variants with polymorphism in the putative drug binding site. In an effort to determine whether the viruses with bevirimat resistant polymorphism also altered their sensitivities to the betulinic acid derivatives that inhibit HIV-1 entry, a series of new betulinic acid entry inhibitors were synthesized and tested for their activities against HIV-1 NL4-3 and NL4-3 variants resistant to bevirimat. The results show that the bevirimat resistant viruses were approximately 5- to10-fold more sensitive to three new glutamine ester derivatives (13, 15 and 38) and A43D in an HIV-1 multi-cycle replication assay. In contrast, the wild type NL4-3 and the bevirimat resistant variants were equally sensitive to the HIV-1 RT inhibitor AZT. In addition, these three new compounds markedly improved microsomal stability compared to A43D.
Co-reporter:Zhao Dang, Andrew Lin, Phong Ho, Dominique Soroka, Kuo-Hsiung Lee, Li Huang, Chin-Ho Chen
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 7) pp:1926-1928
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmcl.2011.02.041
A new class of proteasome inhibitors was synthesized using lithocholic acid as a scaffold. Modification at the C-3 position of lithocholic acid with a series of acid acyl groups yielded compounds with a range of potency on proteasome inhibition. Among them, the phenylene diacetic acid hemiester derivative (13) displayed the most potent proteasome inhibition with IC50 = 1.9 μM. Enzyme kinetic analysis indicates that these lithocholic acid derivatives are noncompetitive inhibitors of the proteasome.
Co-reporter:Zhao Dang ; Weihong Lai ; Keduo Qian ; Phong Ho ; Kuo-Hsiung Lee ; Chin-Ho Chen ;Li Huang
Journal of Medicinal Chemistry 2009 Volume 52(Issue 23) pp:7887-7891
Publication Date(Web):June 15, 2009
DOI:10.1021/jm9004253
We previously reported that [[N-[3β-hydroxyllup-20(29)-en-28-oyl]-7-aminoheptyl]carbamoyl]methane (A43D, 4) was a potent HIV-1 entry inhibitor. However, 4 was inactive against HIV-2 virus, suggesting the structural requirements for targeting these two retroviruses are different. In this study, a series of new betulinic acid derivatives were synthesized, and some of them displayed selective anti-HIV-2 activity at nanomolar concentrations. In comparison to compounds with anti-HIV-1 activity, a shorter C-28 side chain is required for optimal anti-HIV-2 activity.
Co-reporter:Li Huang, Donglei Yu, Phong Ho, Keduo Qian, Kuo-Hsiung Lee, Chin-Ho Chen
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 14) pp:6696-6701
Publication Date(Web):15 July 2008
DOI:10.1016/j.bmc.2008.05.078
This study discovered that glycyrrhetinic acid inhibited the human 20S proteasome at 22.3 μM. Esterification of the C-3 hydroxyl group on glycyrrhetinic acid with various carboxylic acid reagents yielded a series of analogs with marked improved potency. Among the derivatives, glycyrrhetinic acid 3-O-isophthalate (17) was the most potent compound with IC50 of 0.22 μM, which was approximately 100-fold more potent than glycyrrhetinic acid.
Co-reporter:Li Huang, Phong Ho, Kuo-Hsiung Lee, Chin-Ho Chen
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 7) pp:2279-2289
Publication Date(Web):1 April 2006
DOI:10.1016/j.bmc.2005.11.016
Betulinic acid (BA) derivatives with a side chain at C-3 can inhibit HIV-1 maturation. On the other hand, BA derivatives with a side chain at C-28 can block HIV-1 entry. In order to combine the anti-maturation and anti-entry activities in a single molecule, new bi-functional BA derivatives containing side chains at C-3 and C-28 have been synthesized. The most potent compound ([[N-[3β-O-(3′,3′-dimethylsuccinyl)-lup-20(29)-en-28-oyl]-7-aminoheptyl]-carbamoyl]methane) inhibited HIV-1 at an EC50 of 0.0026 μM and was at least 20 times more potent than either the anti-maturation lead compound DSB or the anti-entry lead compound IC9564. This bi-functional BA derivative was active against both HIV entry and maturation. These results suggest that bi-functional BA derivatives with dual mechanisms of action have the potential to become clinically useful for AIDS therapy.Synthesis of betulinic acid derivatives with both anti-HIV entry and anti-HIV maturation activities.
Co-reporter:Li Huang, Phong Ho, Chin-Ho Chen
FEBS Letters (16 October 2007) Volume 581(Issue 25) pp:4955-4959
Publication Date(Web):16 October 2007
DOI:10.1016/j.febslet.2007.09.031
This study discovered that betulinic acid (BA) is a potent proteasome activator that preferentially activates the chymotrypsin-like activity of the proteasome. Chemical modifications can transform BA into proteasome inhibitors. Chemical modifications at the C-3 position of BA resulted in compounds, such as dimethylsuccinyl BA (DSB), with various inhibitory activities against the human 20S proteasome. Interestingly, the proteasomal activation by BA and the inhibitory activity of DSB could be abrogated by introducing a side chain at the C-28 position. In summary, this study discovered a class of small molecules that can either activate or inhibit human proteasome activity depending on side chain modifications.

![16aH-1,6:2,6-Diepoxybenz[7,8]oxireno[5,6]azuleno[8,1-bc]oxacyclotridecin-7,16,16a,17-tetrol, 4-[(benzoyloxy)methyl]eicosahydro-17a-(hydroxymethyl)-](/data/chemimg/597800/60796-70-5.png)
![16aH-1,6:2,6-Diepoxybenz[7,8]oxireno[5,6]azuleno[8,1-bc]oxacyclotridecin-7,16,16a,17-tetrol, 4-[(benzoyloxy)methyl]eicosahydro-17a-(hydroxymethyl)-](/data/chemimg/597800/60796-70-5_b.png)
![4-Piperidinone, 3,5-bis[(4-nitrophenyl)methylene]-1-(1-oxo-2-propen-1-yl)-](/data/chemimg/3753000/330450-45-8.png)
![4-Piperidinone, 3,5-bis[(4-nitrophenyl)methylene]-1-(1-oxo-2-propen-1-yl)-](/data/chemimg/3753000/330450-45-8_b.png)








![(1R)-1,2,3,4,5,6-hexahydro-1,5-methano-8H-pyrido[1,2-a][1,5]diazocin-8-one](http://img.cochemist.com/ccimg/500/486-86-2.png)
![(1R)-1,2,3,4,5,6-hexahydro-1,5-methano-8H-pyrido[1,2-a][1,5]diazocin-8-one](http://img.cochemist.com/ccimg/500/486-86-2_b.png)