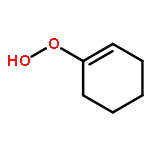
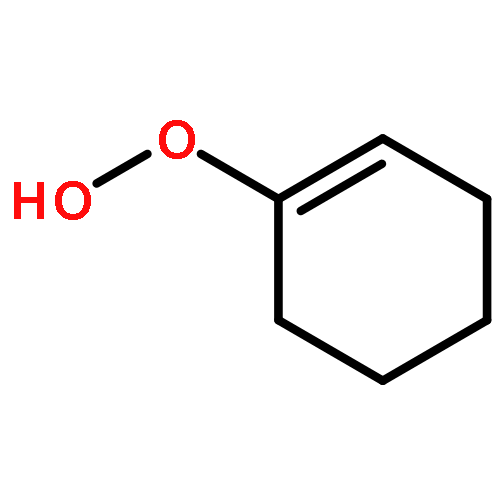
Co-reporter:Luan Nguyen, Lacheng Liu, Solomon Assefa, Christopher Wolverton, William F. Schneider, and Franklin Feng Tao
ACS Catalysis January 6, 2017 Volume 7(Issue 1) pp:664-664
Publication Date(Web):December 6, 2016
DOI:10.1021/acscatal.6b02006
We report direct observation at the atomic scale of the pressure- and temperature-dependent evolution of a model Rh(110) catalyst surface during transient and steady-state CO oxidation, using high-pressure scanning tunneling microscopy (HP-STM) and ambient-pressure X-ray photoelectron spectroscopy (AP-XPS) correlated against density functional theory (DFT) calculations. Rh(110) is susceptible to the well-known missing row (MR) reconstruction. O2 dosing produces a MR structure and an O coverage of 1/2 monolayer (ML), the latter limited by the kinetics of O2 dissociation. In contrast, CO dosing retains the (1 × 1) structure and a CO coverage of 1 ML. We show that CO dosing titrates O from the (2 × 1) structure and that the final surface state is a strong function of temperature. Adsorbed CO accelerates and O inhibits the (2 × 1) to (1 × 1) transition, an effect that can be traced to the influence of the adsorbates on the energy landscape for moving metal atoms from filled to empty rows. During simultaneous dosing of CO and O2, we observed steady-state CO oxidation as well as a transition to the (1 × 1) structure at temperatures more modest than in the titration experiments. This difference may reflect surface heating generated during CO oxidation. At more elevated temperatures the metallic surface transforms to a surface oxide, also active for CO oxidation. Being one of the first examples, these results demonstrate how operando experiment exploration in terms of correlation between surface structure dominated by reaction conditions and activity of a catalytic material and first-principles models can be integrated to disentangle the underlying thermodynamic and kinetic factors that influence the dependence of catalytic activity on surface structure at nano and atomic scales.Keywords: CO oxidation; density functional theory; operando; restructuring; rhodium; STM; XPS;
Co-reporter:Jian Dou;Zaicheng Sun;Adedamola A. Opalade;Nan Wang;Wensheng Fu
Chemical Society Reviews 2017 vol. 46(Issue 7) pp:2001-2027
Publication Date(Web):2017/04/03
DOI:10.1039/C6CS00931J
Chemistry of a catalyst surface during catalysis is crucial for a fundamental understanding of mechanism of a catalytic reaction performed on the catalyst in the gas or liquid phase. Due to the pressure- or molecular density-dependent entropy contribution of gas or liquid phase of the reactants and the potential formation of a catalyst surface during catalysis different from that observed in an ex situ condition, the characterization of the surface of a catalyst under reaction conditions and during catalysis can be significant and even necessary for understanding the catalytic mechanism at a molecular level. Electron-based analytical techniques are challenging for studying catalyst nanoparticles in the gas or liquid phase although they are necessary techniques to employ. Instrumentation and further development of these electron-based techniques have now made in situ/operando studies of catalysts possible. New insights into the chemistry and structure of catalyst nanoparticles have been uncovered over the last decades. Herein, the origin of the differences between ex situ and in situ/operando studies of catalysts, and the technical challenges faced as well as the corresponding instrumentation and innovations utilized for characterizing catalysts under reaction conditions and during catalysis, are discussed. The restructuring of catalyst surfaces driven by the pressure of reactant(s) around a catalyst, restructuring in reactant(s) driven by reaction temperature and restructuring during catalysis are also reviewed herein. The remaining challenges and possible solutions are briefly discussed.
Co-reporter:Jian Dou;Yu Tang;Luan Nguyen;Xiao Tong;Prem S. Thapa
Catalysis Letters 2017 Volume 147( Issue 2) pp:442-452
Publication Date(Web):2017 February
DOI:10.1007/s10562-016-1883-6
Nanoporous gold with minor silver content has been identified as a new type of gold based catalyst for selective oxidation of cyclohexene with molecular oxygen in liquid. By oxidation of the leached nanoporous gold foils in ozone, the minor silver content was oxidized to form silver oxide nanoclusters on the surface of nanoporous gold. With further treatment in methanol, the surface silver oxide was reduced and surface alloy was formed on gold ligaments. Both nanoporous gold treated with ozone only and the one with ozone and then methanol are very active for selective oxidation of cyclohexene with molecular oxygen in liquid of cyclohexene with a turn-over-frequency (TOF) of 0.55–0.99 molecules per surface Au atom per second under a solvent-free and initiator- free condition. The total selectivity for production of 2-cyclohexene-1-one, 2-cyclohexene-1-ol, and cyclohexene oxide was increased from 57.5 % to 80.8 % by an additional treatment of nanoporous gold in methanol after activation in zone. The correlation of catalytic selectivity for the production of the three products and corresponding surface chemistry of ligament suggests that (1) the formed Au–Ag alloy surface is favorable for the formation of 2-cyclohexen-1-one, 2-cyclohexene-1-ol, and cyclohexene oxide and (2) the surface silver oxide is favorable for the production of cyclohexenyl hydroperoxide.
Co-reporter:Franklin (Feng) Tao and Peter A. Crozier
Chemical Reviews 2016 Volume 116(Issue 6) pp:3487
Publication Date(Web):March 9, 2016
DOI:10.1021/cr5002657
Heterogeneous catalysis is a chemical process performed at a solid–gas or solid–liquid interface. Direct participation of catalyst atoms in this chemical process determines the significance of the surface structure of a catalyst in a fundamental understanding of such a chemical process at a molecular level. High-pressure scanning tunneling microscopy (HP-STM) and environmental transmission electron microscopy (ETEM) have been used to observe catalyst structure in the last few decades. In this review, instrumentation for the two in situ/operando techniques and scientific findings on catalyst structures under reaction conditions and during catalysis are discussed with the following objectives: (1) to present the fundamental aspects of in situ/operando studies of catalysts; (2) to interpret the observed restructurings of catalyst and evolution of catalyst structures; (3) to explore how HP-STM and ETEM can be synergistically used to reveal structural details under reaction conditions and during catalysis; and (4) to discuss the future challenges and prospects of atomic-scale observation of catalysts in understanding of heterogeneous catalysis. This Review focuses on the development of HP-STM and ETEM, the in situ/operando characterizations of catalyst structures with them, and the integration of the two structural analytical techniques for fundamentally understanding catalysis.
Co-reporter:Shaowen Cao, Franklin (Feng) Tao, Yu Tang, Yuting Li and Jiaguo Yu
Chemical Society Reviews 2016 vol. 45(Issue 17) pp:4747-4765
Publication Date(Web):08 Jun 2016
DOI:10.1039/C6CS00094K
Heterogeneous catalysis is one of the most important chemical processes of various industries performed on catalyst nanoparticles with different sizes or/and shapes. In the past two decades, the catalytic performances of different catalytic reactions on nanoparticles of metals and oxides with well controlled sizes or shapes have been extensively studied thanks to the spectacular advances in syntheses of nanomaterials of metals and oxides. This review discussed the size and shape effects of catalyst particles on catalytic activity and selectivity of reactions performed at solid–gas or solid–liquid interfaces with a purpose of establishing correlations of size- and shape-dependent chemical and structural factors of surface of a catalyst with the corresponding catalytic performances toward understanding of catalysis at a molecular level.
Co-reporter:Franklin Feng Tao, Luan Nguyen, Shiran Zhang, Yuanyuan Li, Yu Tang, Lei Zhang, Anatoly I. Frenkel, Younan Xia, and Miquel Salmeron
Nano Letters 2016 Volume 16(Issue 8) pp:5001-5009
Publication Date(Web):June 21, 2016
DOI:10.1021/acs.nanolett.6b01718
Heterogeneous catalysis occurs at the interface between a solid catalyst and the reactants. The structure of metal catalyst nanoparticles at the metal–gas interface is a key factor that determines catalytic selectivity and activity. Here we report that second-generation nanoclusters are formed on the initial catalyst nanoparticles as a result of interaction with the reactant molecules when the nanoparticles are in a gas phase at Torr pressure or higher. The formation of the second-generation nanoclusters is manifested by a decrease of the average coordination number of the metal atoms and a shift of their core level energies in the presence of gases. The formation of second-generation nanoclusters increases the number of undercoordinated sites, which are the most active for catalysis in many cases.
Co-reporter:Juanjuan Liu, Shiran Zhang, Yan Zhou, Victor Fung, Luan Nguyen, De-en Jiang, Wenjie Shen, Jie Fan, and Franklin Feng Tao
ACS Catalysis 2016 Volume 6(Issue 7) pp:4218
Publication Date(Web):June 6, 2016
DOI:10.1021/acscatal.5b02900
Catalytic selectivity for producing an ideal product is a key topic for chemical transformations through heterogeneous catalysis. Tuning catalytic selectivity by integrating the second metal to form an alloy has been well demonstrated in the literature. Here we report a method to tune catalytic selectivity in oxidative catalysis on another category of heterogeneous catalysts, transition-metal oxides. By choosing the oxidative dehydrogenation (ODH) of ethane to ethylene as a probe reaction, we demonstrated that doping nonmetallic atoms to the surface lattice of catalyst of a transition-metal oxide can enhance catalytic selectivity through suppression of complete oxidation of the reactant molecules. Catalysts of Co3O4 with doped silicon atoms (Six-Co3O4) maintaining the spinel structure of pure Co3O4 exhibit much higher selectivity for the production of ethylene through ODH of ethane in comparison to pure Co3O4 at 600 °C by 40%. The suppression of activity of surface lattice oxygen atoms was evidenced by the observation that the surface lattice oxygen atoms of Six-Co3O4 cannot exchange oxygen atoms with gas-phase oxygen at low temperatures while pure Co3O4 can. The difference in releasing surface lattice oxygen atoms and dissociating molecular oxygen between pure Co3O4 and Six-Co3O4 was supported by DFT calculations. The calculated activation barriers for dissociation of molecular O2 and energy barriers for hopping surface oxygen vacancies of Six-Co3O4 are obviously higher than those of pure Co3O4, respectively. These experimental exploration and computational studies established a correlation between increase of catalytic selectivity and suppression of the activity of surface lattice oxygen atoms/oxygen vacancies. This correlation suggests an approach for increasing the catalytic selectivity of oxidative catalysis through suppressing activity of surface lattice oxygen atoms/vacancies via doping atoms of a nonmetallic element. This new approach was further confirmed by the observed higher catalytic selectivity for production of ethylene on Ge0.2-Co3O4 in comparison to pure Co3O4.Keywords: catalytic selectivity; cobalt oxide; ethane; ethylene; oxidative dehydrogenation; oxygen vacancies; surface lattice oxygen
Co-reporter:Luan Nguyen, Shiran Zhang, Lei Wang, Yuanyuan Li, Hideto Yoshida, Anitha Patlolla, Seiji Takeda, Anatoly I. Frenkel, and Franklin (Feng) Tao
ACS Catalysis 2016 Volume 6(Issue 2) pp:840
Publication Date(Web):December 10, 2015
DOI:10.1021/acscatal.5b00842
The bimetallic catalyst has been one of the main categories of heterogeneous catalysts for chemical production and energy transformation. Isolation of the continuously packed bimetallic sites of a bimetallic catalyst forms singly dispersed bimetallic sites which have distinctly different chemical environment and electronic state and thus exhibit a different catalytic performance. Two types of catalysts consisting of singly dispersed bimetallic sites Pt1Com or Pd1Con (m and n are the average coordination numbers of Co to a Pt or Pd atom) were prepared through a deposition or impregnation with a following controlled calcination and reduction to form Pt1Com or Pd1Con sites. These bimetallic sites are separately anchored on a nonmetallic support. Each site only consists of a few metal atoms. Single dispersions of these isolated bimetallic sites were identified with scanning transmission electron microscopy. Extended X-ray absorption fine structure spectroscopy (EXAFS) revealed the chemical bonding of single atom Pt1 (or Pd1) to Co atoms and thus confirmed the formation of bimetallic sites, Pt1Com and Pd1Con. Reduction of NO with H2 was used as a probing reaction to test the catalytic performance on this type of catalyst. Selectivity in reducing nitric oxide to N2 on Pt1Com at 150 °C is 98%. Pd1Con is active for reduction of NO with a selectivity of 98% at 250 °C. In situ studies of surface chemistry with ambient-pressure X-ray photoelectron spectroscopy and coordination environment of Pt and Pd atoms with EXAFS showed that chemical state and coordination environment of Pt1Com and Pd1Con remain during catalysis up to 250 and 300 °C, respectively. The correlation of surface chemistries and structures of these catalysts with their corresponding catalytic activities and selectivities suggests a method to develop new bimetallic catalysts and a new type of single site catalysts.Keywords: AP-XPS; bimetallic; EXAFS; in-situ and operando; single site catalysis
Co-reporter:Longhui Nie, Jiaguo Yu, Mietek Jaroniec and Franklin Feng Tao
Catalysis Science & Technology 2016 vol. 6(Issue 11) pp:3649-3669
Publication Date(Web):14 Mar 2016
DOI:10.1039/C6CY00062B
Formaldehyde (HCHO) is one of the major pollutants in indoor air and long-term exposure to HCHO even at very low concentrations is harmful and may cause health problems including nasal tumors and skin irritation. Removal of HCHO is necessary to improve the quality of indoor air. Catalytic oxidative decomposition of HCHO at room temperature is regarded as the most promising strategy for the removal of HCHO because it is environmentally friendly and energy-saving. In this review, the reported catalytic materials for room-temperature catalytic oxidative decomposition of HCHO are discussed. In addition, the development and performance of catalysts for HCHO oxidative decomposition, the mechanism of this catalytic process, the surface chemistry and structure factors influencing catalytic performances, existing challenges in the development of catalysts with low cost and high activity and perspectives for important topics of future research in this area were reviewed.
Co-reporter:Shiran Zhang, Junjun Shan, Longhui Nie, Luan Nguyen, Zili Wu, Franklin (Feng) Tao
Surface Science 2016 Volume 648() pp:156-162
Publication Date(Web):June 2016
DOI:10.1016/j.susc.2015.12.011
NiFe2O4 with an inverse spinel structure exhibits high activity for a complete oxidation of methane at 400 °C–425 °C and a higher temperature. The surface of the catalyst and its adsorbates were well characterized with ambient pressure X-ray photoelectron spectroscopy (AP-XPS) and in situ infrared spectroscopy (IR). In situ studies of the surface of NiFe2O4 using AP-XPS suggest the formation of methoxy-like and formate-like intermediates at a temperature lower than 200 °C, supported by the observed vibrational signatures in in situ IR studies. Evolutions of C1s photoemission features and the nominal atomic ratios of C/(Ni + Fe) of the catalyst surface suggest that the formate-like intermediate is transformed to product molecules CO2 and H2O in the temperature range of 250–300 °C. In situ studies suggest the formation of a spectator, − OlatticeCH2Olattice −. It strongly bonds to surface through CO bonds and cannot be activated even at 400 °C.
Co-reporter:Hyuntae Sohn
The Journal of Physical Chemistry C 2016 Volume 120(Issue 27) pp:14631-14642
Publication Date(Web):June 17, 2016
DOI:10.1021/acs.jpcc.6b02490
The surface and bulk reduction characteristics of bare ceria and ceria with supported cobalt nanoparticles were investigated under ethanol steam reforming conditions using AP-XPS and XANES techniques. Ceria particles were prepared in two different particle sizes, one in nano and the other in micron size (termed CeO2-NP and CeO2-MP), with average particle sizes of 4 and 120 nm, respectively. It was found that particle size affects surface reducibility of ceria particles; smaller particle size leads to a higher extent of surface reduction. Supported cobalt nanoparticles have a significant effect on the surface reducibility of both CeO2-NP and CeO2-MP. Compared to bare ceria particles, the presence of fully oxidized cobalt nanoparticles on the surface of ceria support retards surface reducibility of ceria since reduction of the cobalt oxide phases (Co3O4 and CoO) takes precedence over that of ceria. The degree of reduction of the cobalt phase during ethanol steam reforming determines the effect of cobalt on the reduction process of ceria, i.e., whether it retards or facilitates the reduction of ceria support. AP-XPS studies show that the surface of cobalt nanoparticles consists of both metallic Co and CoOx. The reduction of the surface region of CoOx to metallic Co forms a metallic Co-based shell and CoOx-based core. The anchor of metallic Co on CoOx make metallic Co shell well dispersed on CeO2 without sintering. In addition, the reforming reaction takes place primarily at the interface of metallic Co and CeO2. The much larger difference between Co/CeO2-NP and Co/CeO2-MP than the difference between CeO2-NP and CeO2-MP suggests the significance of metallic Co in catalyzing the reforming reaction, although bare ceria support shows some dehydration activity in its own right.
Co-reporter:Jun-jun Shan;Luan Nguyen;Shiran Zhang;Franklin-Feng Tao
Catalysis Letters 2015 Volume 145( Issue 8) pp:1571-1580
Publication Date(Web):2015 August
DOI:10.1007/s10562-015-1549-9
Low temperature water–gas shift (WGS) catalysts, Pd nanoparticles supported on α-MnO2 nanorods termed Pd/α-MnO2 and Pt nanoparticles supported on α-MnO2 nanorods termed Pt/α-MnO2 were synthesized by introducing Pd or Pt precursor to well-prepared α-MnO2 nanorods through precipitation deposition with a following annealing at 300 °C. They are quite active for WGS in the temperature range of 140–350 °C. Activation energies for WGS on Pd/α-MnO2 and Pt/α-MnO2 are 45.3 and 56.4 kJ/mol respectively, comparable to precious metal supported on CeO2 and TiO2 for WGS. Surface chemistries of the two catalysts during WGS were tracked with ambient pressure X-ray photoelectron spectroscopy. Different from the preservation of the surface and bulk phase of other oxide support such as CeO2, TiO2 in CeO2- or TiO2-based WGS catalysts, both surface and bulk of α-MnO2 nanorods of Pd/α-MnO2 and Pt/α-MnO2 are transited to MnO during WGS. In-situ studies identified oxygen vacancies of the formed MnO support during WGS and the metallic state of Pd and Pt nanoparticles supported on the nonstoichiometric MnO.
Co-reporter:Yu Tang, Luan Nguyen, Yuting Li, Nan Wang, Franklin (Feng) Tao
Current Opinion in Chemical Engineering (May 2016) Volume 12() pp:52-61
Publication Date(Web):1 May 2016
DOI:10.1016/j.coche.2016.02.007
•Active surface of a catalyst could be formed in a pretreatment or/and a reaction with one or more reactants.•Restructuring is one important surface process of some catalysts.•A correlation between a catalyst surface and its corresponding catalytic performance can be established upon the identification of active surface.Chemistry and structure of surface of a catalyst under a reaction condition is the crucial information for understanding catalytic mechanism since in many cases an authentic, active surface catalyzing a catalytic reaction is formed in a pretreatment or/and in a reaction between nominal catalyst and reactants. Ambient pressure X-ray photoelectron spectroscopy can be used to track surface of a catalyst under a reaction condition as the instrumentations in the last a few decades have made characterization of catalyst surface in a gas phase at Torr pressure or higher possible. It can characterize surface chemistry of a catalyst including surface composition, surface phase and surface oxygen vacancies and other information under a reaction condition and track their evolutions when the reaction condition is changed to another.
Co-reporter:Jian Dou, Franklin (Feng) Tao
Applied Catalysis A: General (5 January 2017) Volume 529() pp:
Publication Date(Web):5 January 2017
DOI:10.1016/j.apcata.2016.10.010
•MoO3@np-Au catalyst is active for selective oxidation of cyclohexene with molecular oxygen.•MoO3@np-Au catalysts exhibited high selectivity of 58%–73% for cyclohexene oxide at conversion of 4%–11% of cyclohexene.•MoO3@np-Au catalyst behaves as a bi-functional catalyst.Selective oxidation of olefins to desired products such as epoxides is highly demanded in chemical industry. It remains challenging to achieve high selectivity for production of epoxides over heterogeneous catalysts through using molecular oxygen as the oxidant. Nanoporous reverse catalysts (MoO3@np-Au) consisting of pure nanoporous gold (np-Au) and MoO3 nanoparticles anchored on Au ligaments were synthesized for selective oxidation of cyclohexene with molecular oxygen. By controlling the loading of molybdenum and thermal treatment condition, MoO3 nanoparticles with size of ∼5 nm were uniformly anchored on the surface of gold ligaments (30–50 nm) of pure nanoporous gold (np-Au). These synthesized MoO3@np-Au catalysts exhibited high selectivity of 58%–73% for production of cyclohexene oxide at conversion of 4%–11% of cyclohexene by using molecular oxygen as the oxidant. Compared to MoO3@np-Au, the selectivity for the production of cyclohexene oxide on pure np-Au catalyst is only 6% under the same catalytic condition as that on MoO3@np-Au. The observed high selectivity for production of cyclohexene oxide on MoO3@np-Au can be rationalized with a bi-functional mechanism of a reverse metal/oxide catalyst. The in-situ formed surface molybdenum oxo-peroxo species are suggested to be responsible for selective oxidation of cyclohexene to cyclohexene oxide, while the MoO3/Au interface activates molecular oxygen to regenerate the molybdenum oxo-peroxo active centers.
Co-reporter:Shaowen Cao, Franklin (Feng) Tao, Yu Tang, Yuting Li and Jiaguo Yu
Chemical Society Reviews 2016 - vol. 45(Issue 17) pp:NaN4765-4765
Publication Date(Web):2016/06/08
DOI:10.1039/C6CS00094K
Heterogeneous catalysis is one of the most important chemical processes of various industries performed on catalyst nanoparticles with different sizes or/and shapes. In the past two decades, the catalytic performances of different catalytic reactions on nanoparticles of metals and oxides with well controlled sizes or shapes have been extensively studied thanks to the spectacular advances in syntheses of nanomaterials of metals and oxides. This review discussed the size and shape effects of catalyst particles on catalytic activity and selectivity of reactions performed at solid–gas or solid–liquid interfaces with a purpose of establishing correlations of size- and shape-dependent chemical and structural factors of surface of a catalyst with the corresponding catalytic performances toward understanding of catalysis at a molecular level.
Co-reporter:Longhui Nie, Jiaguo Yu, Mietek Jaroniec and Franklin Feng Tao
Catalysis Science & Technology (2011-Present) 2016 - vol. 6(Issue 11) pp:NaN3669-3669
Publication Date(Web):2016/03/14
DOI:10.1039/C6CY00062B
Formaldehyde (HCHO) is one of the major pollutants in indoor air and long-term exposure to HCHO even at very low concentrations is harmful and may cause health problems including nasal tumors and skin irritation. Removal of HCHO is necessary to improve the quality of indoor air. Catalytic oxidative decomposition of HCHO at room temperature is regarded as the most promising strategy for the removal of HCHO because it is environmentally friendly and energy-saving. In this review, the reported catalytic materials for room-temperature catalytic oxidative decomposition of HCHO are discussed. In addition, the development and performance of catalysts for HCHO oxidative decomposition, the mechanism of this catalytic process, the surface chemistry and structure factors influencing catalytic performances, existing challenges in the development of catalysts with low cost and high activity and perspectives for important topics of future research in this area were reviewed.
Co-reporter:Jian Dou, Zaicheng Sun, Adedamola A. Opalade, Nan Wang, Wensheng Fu and Franklin (Feng) Tao
Chemical Society Reviews 2017 - vol. 46(Issue 7) pp:NaN2027-2027
Publication Date(Web):2017/03/30
DOI:10.1039/C6CS00931J
Chemistry of a catalyst surface during catalysis is crucial for a fundamental understanding of mechanism of a catalytic reaction performed on the catalyst in the gas or liquid phase. Due to the pressure- or molecular density-dependent entropy contribution of gas or liquid phase of the reactants and the potential formation of a catalyst surface during catalysis different from that observed in an ex situ condition, the characterization of the surface of a catalyst under reaction conditions and during catalysis can be significant and even necessary for understanding the catalytic mechanism at a molecular level. Electron-based analytical techniques are challenging for studying catalyst nanoparticles in the gas or liquid phase although they are necessary techniques to employ. Instrumentation and further development of these electron-based techniques have now made in situ/operando studies of catalysts possible. New insights into the chemistry and structure of catalyst nanoparticles have been uncovered over the last decades. Herein, the origin of the differences between ex situ and in situ/operando studies of catalysts, and the technical challenges faced as well as the corresponding instrumentation and innovations utilized for characterizing catalysts under reaction conditions and during catalysis, are discussed. The restructuring of catalyst surfaces driven by the pressure of reactant(s) around a catalyst, restructuring in reactant(s) driven by reaction temperature and restructuring during catalysis are also reviewed herein. The remaining challenges and possible solutions are briefly discussed.
.jpg)