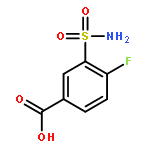
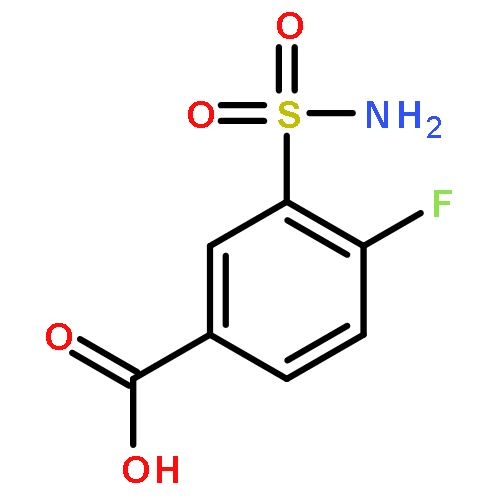
Co-reporter:Lin-Fu Liang, Ting Wang, You-Sheng Cai, Wen-Fei He, Peng Sun, Yu-Fen Li, Qi Huang, Orazio Taglialatela-Scafati, He-Yao Wang, Yue-Wei Guo
European Journal of Medicinal Chemistry 2014 Volume 79() pp:290-297
Publication Date(Web):22 May 2014
DOI:10.1016/j.ejmech.2014.04.003
•Eight new brominated polyunsaturated lipids (3–10) have been characterized.•The reported compounds represent a new class of pancreatic lipase (PL) inhibitors.•Compound 14 exhibited the strongest inhibitory activity against PL.•14 strongly suppressed serum triglyceride elevation in olive oil-loaded mice.•No signs related death or abnormality were observed after 14 administration.Chemical analysis of the Chinese marine sponge Xestospongia testudinaria afforded a library of brominated polyunsaturated lipids including eight new compounds, named xestonarienes A–H (3–10) and thirteen known analogues (11–23). The structures of the new compounds were elucidated by detailed spectroscopic analysis and by comparison with literature data. The isolated lipids were evaluated for their inhibitory activity against pancreatic lipase (PL), an essential enzyme for efficient fat digestion and the major metabolite, 14, exhibited a marked inhibitory activity (IC50 = 3.11 μM), similar to that of the positive control Orlistat (IC50 = 0.78 μM). The preliminary structure–activity relationships on the series of compounds clearly evidenced that a terminal (E)-enyne functionality, a diyne within the chain, and methyl ester group are all key functional groups for the activity of this class of PL inhibitors. Further biological investigation on compound 14 revealed a significant decrease in the plasma triglyceride level following an oral lipid challenge in C57BLKS/J male mice. Acute toxicology study demonstrated that compound 14 was non-toxic up to 1600 mg/kg p.o in mice. This is the first report of the PL inhibitory activity for brominated polyunsaturated lipids and the obtained results qualify compound 14 as a potent and bioavailable drug candidate for a mild and safe treatment to prevent and reduce obesity.Methyl xestospongic ester (14) exhibited a marked inhibitory activity against pancreatic lipase, representing a new structural class of inhibitors. In the acute toxicity study, no signs related death or abnormality were observed after compound 14 administration.
Co-reporter:Bo Li, Gaihong Wang, Zhijian Xu, Yong Zhang, Xiangui Huang, Bubing Zeng, Kaixian Chen, Jiye Shi, Heyao Wang, Weiliang Zhu
European Journal of Medicinal Chemistry 2014 Volume 77() pp:204-210
Publication Date(Web):22 April 2014
DOI:10.1016/j.ejmech.2014.03.008
•Several N-substituted isoquinolones were obtained via [2,3] or [3,3] rearrangement.•4h and 4j showed strong antitumor activities close to vemurafenib and sorafenib.•The unusual [2,3] rearrangement is a novel rearrangement observed on lactams.The present study discovers multiple N-substituted 3-arylisoquinolone derivatives as antitumor agents originating from O-substituted 3-arylisoquinolines via [2,3] or [3,3] rearrangement. The current [2,3] rearrangement of epoxy or acetal O-substituents converting to diol or alcohol N-substituents can be promoted by silica gel or by diluted hydrochloric acid, which is distinct from previously reported [2,3] rearrangements. Some of the derivatives displayed comparable or even stronger cytotoxicity than sorafenib and vemurafenib on HCT116 colon carcinoma and A375 melanoma cell lines. Therefore, the rearrangement via intramolecular carbon-oxygen bond cleavage and carbon-nitrogen bond formation should be a useful approach for developing novel anticancer drugs derived from isoquinolones.Several N-substituted isoquinolones were obtained via [2,3] or [3,3] rearrangement, among which, 4h and 4j showed strong antitumor activities close to vemurafenib and sorafenib.
Co-reporter:Ting Wang, Peng Sun, Liang Chen, Qi Huang, Kaixian Chen, Qi Jia, Yiming Li, and Heyao Wang
Journal of Agricultural and Food Chemistry 2014 Volume 62(Issue 22) pp:5038-5045
Publication Date(Web):May 12, 2014
DOI:10.1021/jf500387d
In previous studies, A-type procyanidin oligomers isolated from Cinnamomum tamala were proved to possess antidiabetic effect and protect pancreatic β-cells in vivo. The aim of this study was to unveil the mechanisms of protecting pancreatic β-cells from palmitic acid-induced apoptosis by cinnamtannin D-1 (CD1), one of the main A-type procyanidin oligomers in C. tamala. CD1 was discovered to dose-dependently reduce palmitic acid- or H2O2-induced apoptosis and oxidative stress in INS-1 cells, MIN6 cells, and primary cultured murine islets. Moreover, CD1 could reverse palmitic acid-induced dysfunction of glucose-stimulated insulin secretion in primary cultured islets. These results indicate that reduction of apoptosis and oxidative stress might account for the protection effect of CD1, which provided a better understanding of the mechanisms of the antidiabetic effects of procyanidin oligomers.
Co-reporter:Xiaodong Zhang;Guirui Yan;Jun Ji;Jingwei Wu;Xiaoyun Sun;Jingshan Shen;Hualiang Jiang
Journal of Cellular Biochemistry 2012 Volume 113( Issue 8) pp:2738-2743
Publication Date(Web):
DOI:10.1002/jcb.24147
Abstract
PDE inhibitors could increase cellular cGMP levels and are used to treat erectile dysfunction as well as pulmonary arterial hypertension. cGMP production was reported to be necessary for UVB-induced melanin synthesis, however, the effect of PDE5 inhibitor on melanin synthesis has not been examined. We found that PDE5 inhibitor (sildenafil or vardenafil) and the cGMP analog 8-CPT-cGMP stimulated CREB phosphorylation, leading to increased tyrosinase expression and melanin synthesis, which was counteracted by KT5823, a selective cGMP-dependent protein kinase (PKG) inhibitor. However, KT5823 did not affect cAMP-elevating agent-mediated melanin synthesis, indicating that KT5823 selectively inhibited cGMP-induced melanin synthesis. This is the first study to find that PDE5 inhibitor can promote melanin synthesis and reveal that PKG-dependent CREB phosphorylation and tyrosinase expression is involved in cGMP-induced melanin synthesis. Our results suggest that PDE5 inhibitor may be beneficial for the treatment of hypopigmentation diseases. J. Cell. Biochem. 113: 2738–2743, 2012. © 2012 Wiley Periodicals, Inc.
Co-reporter:Jinwei Wu;Peng Sun;Xiaodong Zhang;Hong Liu;Hualiang Jiang;Weiliang Zhu
Journal of Cellular Biochemistry 2012 Volume 113( Issue 4) pp:1152-1158
Publication Date(Web):
DOI:10.1002/jcb.23450
Abstract
Chronic exposure to elevated concentration of free fatty acids (FFA) has been verified to induce endoplasmic reticulum (ER) stress, which leads to pancreatic β-cell apoptosis. As one of the medium and long chain FFA receptors, GPR40 is highly expressed in pancreatic β cells, mediates both acute and chronic effects of FFA on β-cell function, but the role of GPR40 in FFA-induced β-cell apoptosis remains unclear. In this study, we investigated the possible effects of GPR40 in palmitate-induced MIN6 β-cell apoptosis, and found that DC260126, a novel small molecular antagonist of GPR40, could protect MIN6 β cells from palmitate-induced ER stress and apoptosis. Similar results were observed in GPR40-deficient MIN6 cells, indicating that palmitate-induced β-cell apoptosis is at least partially dependent on ER stress pathway via GRP40. J. Cell. Biochem. 113: 1152–1158, 2012. © 2011 Wiley Periodicals, Inc.
Co-reporter:Liang Chen, Peng Sun, Ting Wang, Kaixian Chen, Qi Jia, Heyao Wang, and Yiming Li
Journal of Agricultural and Food Chemistry 2012 Volume 60(Issue 36) pp:9144-9150
Publication Date(Web):August 25, 2012
DOI:10.1021/jf3024535
The procyanidin oligomers are thought to be responsible for the antidiabetic activity of cinnamon. To investigate the hypoglycemic effects of different procyanidin oligomer types, the procyanidin oligomer-rich extracts were prepared from two different cinnamon species. Using high-performance liquid chromatography with purified procyanidin oligomers as reference compounds, we found that the Cinnamomum cassia extract (CC-E) and Cinnamomum tamala extract (CT-E) were rich in B- and A-type procyanidin oligomers, respectively. In the experiment, 8-week-old diabetic (db/db) mice were gavaged with CC-E and CT-E (both 200 mg/kg per day) for 4 weeks. Both CC-E and CT-E exhibited antidiabetic effects. Moreover, histopathological studies of the pancreas, liver, and adipose tissue showed that CC-E promoted lipid accumulation in the adipose tissue and liver, whereas CT-E mainly improved the insulin concentration in the blood and pancreas.
Co-reporter:Zhijian Xu, Guirui Yan, Gaihong Wang, Bo Li, Jianming Zhu, Peng Sun, Xiaodong Zhang, Cheng Luo, Heyao Wang, Weiliang Zhu
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 17) pp:5428-5437
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmcl.2012.07.039
In this study for searching novel B-RafV600E inhibitors, pharmacophore-based virtual screening identified 1 as a hit bearing 5-benzylidene-2-thioxodihydropyrimidine-4,6(1H,5H)-dione. Based on 1, scaffold hopping inspired by molecular docking discovered 5-(furan-2-ylmethylene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione as a new and better scaffold. Substructure search with the new scaffold identified 28 active compounds, among which 12 compounds (42.9%) showed IC50 less than 1 μM. Especially, compound 3o, which is 10-fold more potent than the hit 1, is a potent inhibitor comparable to that of the marketed drug vemurafenib.
Co-reporter:Xiao Hu, Jin-Wei Wu, Xiao-Dong Zhang, Qin-Shi Zhao, Jian-Ming Huang, He-Yao Wang, and Ai-Jun Hou
Journal of Natural Products 2011 Volume 74(Issue 4) pp:816-824
Publication Date(Web):March 14, 2011
DOI:10.1021/np100907d
Ten new isoprenylated flavonoids, nigrasins A−J (1−10), and three known compounds were isolated from the twigs of Morus nigra. Compounds 8 and 9 promoted adipogenesis, characterized by increased lipid droplet and triglyceride content in 3T3L1 cells, and induced up-regulation of the expression of adipocyte-specific genes, aP2 and GLUT4.
Co-reporter:Haiyan Cai, Guirui Yan, Xiaodong Zhang, Olena Gorbenko, Heyao Wang, Weiliang Zhu
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 12) pp:3675-3679
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmcl.2010.04.095
In this study, a series of small molecule inhibitors of human FABP4 were identified through virtual screening. Compound 1 is the most potent hit against FABP4 with a selectivity of more than 144-fold preferences over human FABP3. In addition, MD simulation and mutation studies revealed key residues for inhibitory potency and selectivity, which provides a guideline for further drug design against obesity, diabetes and atherosclerosis.Compound 1 was identified as an inhibitor of human FABP4 with an IC50 of 13.5 μM and it showed a selectivity more than 144-fold over human FABP3.
Co-reporter:Xu Sun;Xiao-Dong Zhang;Gang Cheng;You-Hong Hu
Molecular and Cellular Biochemistry 2009 Volume 330( Issue 1-2) pp:
Publication Date(Web):2009 October
DOI:10.1007/s11010-009-0131-4
Hepatic stellate cells (HSCs) play an important role in the development of hepatic fibrosis. Heat shock protein 90 (Hsp90) is essential for the maturation and activity of a varied group of proteins involved in signal transduction and cell cycle regulation. In this study, we found that two Hsp90 inhibitors, VER-49009 and its analog VER-49009M, inhibited the proliferation of hepatic stellate cell line CFSC cells, and both of them induced G2 phase arrest in CFSC cells. Akt expression was decreased by the treatment of Hsp90 inhibitors in CFSC cells. Based on these findings, we propose that the inhibition of Hsp90 might be a rational approach in the prevention of liver fibrosis.
Co-reporter:YONGGANG WANG;FENGRONG ZHU;FUSEN HAN
Journal of Food Biochemistry 2008 Volume 32( Issue 5) pp:654-671
Publication Date(Web):
DOI:10.1111/j.1745-4514.2008.00190.x
ABSTRACT
Protamine, derived from fish milt, which is normally discarded as an industrial by-product in the process of fish plant, was hydrolyzed with pancreatin. Salmon protamine hydrolysate was found to possess antioxidative activity against hydroxyl, 2,2-diphenyl-1-picrylhydrazyl and superoxide anion radicals. Through consecutive chromatographic methods including size exclusion, ion exchange chromatography and reverse-phase high performance liquid chromatography (HPLC), a series of peptide fractions with high antioxidative activities were obtained. Peptide with highest antioxidative activity was identified to be Pro-Arg matching 1–2 and 16–17 residues of the salmon protamine by electrospray ionization mass spectrometry and database search. These results provided sufficient and scientific information for the marketing of health food supplemented with protamine hydrolysate and added further support for the wide applications of fish milt.
PRACTICAL APPLICATIONS
Protamine could be used to produce protamine hydrolysate with antioxidative activity. There are few reports on protamine hydrolysate as natural health food, especially its bioactive fraction. This work reports isolation and characterization of antioxidative peptides from protamine hydrolysate, which was derived from salmon milt. It is beneficial to utilize fish milt, which is readily available in large quantities and at low prices. Furthermore, the use of protamine hydrolysate in health food is gaining much more interest.

![N-[3-[(5-Chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)carbonyl]-2,4-difluorophenyl]-1-propanesulfonamide](http://img.cochemist.com/ccimg/918600/918505-84-7.png)
![N-[3-[(5-Chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)carbonyl]-2,4-difluorophenyl]-1-propanesulfonamide](http://img.cochemist.com/ccimg/918600/918505-84-7_b.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1.png)
![N-(3-(5-(4-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridine-3-carbonyl)-2,4-difluorophenyl)propane-1-sulfonamide](http://img.cochemist.com/ccimg/918600/918504-65-1_b.png)





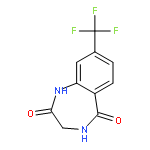
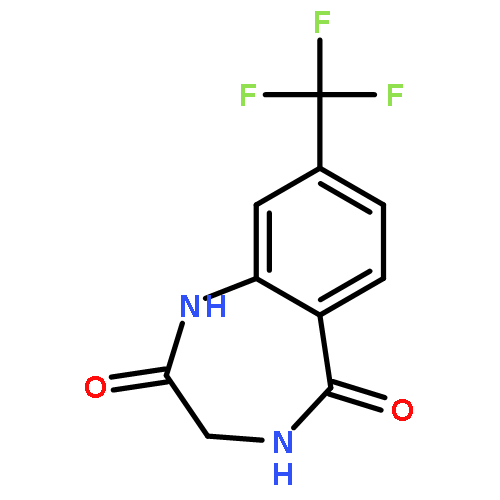
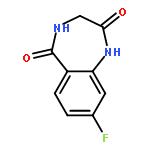
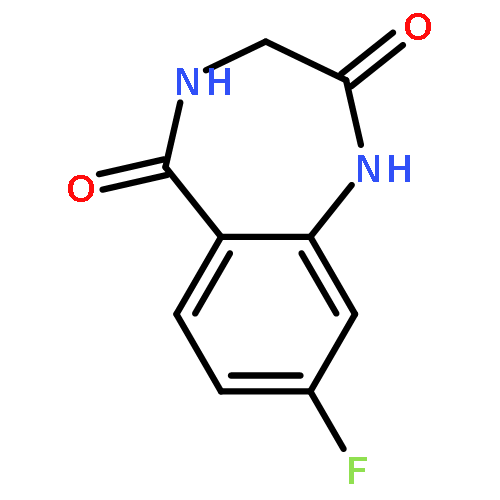
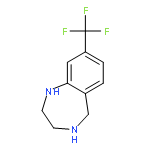
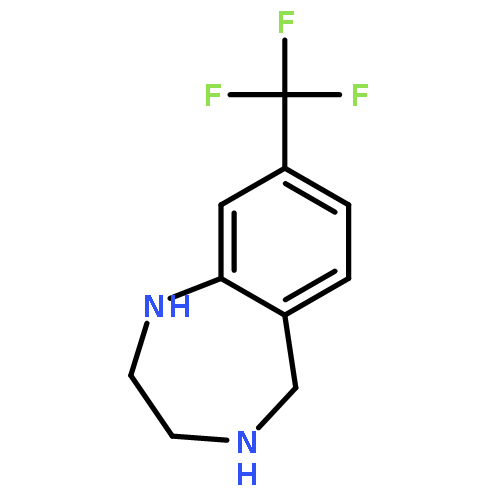
![8-Fluoro-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine](http://img.cochemist.com/ccimg/621000/620948-83-6.png)
![8-Fluoro-2,3,4,5-tetrahydro-1H-benzo[e][1,4]diazepine](http://img.cochemist.com/ccimg/621000/620948-83-6_b.png)
![1H-Benzimidazole, 1-[[2,4,6-tris(1-methylethyl)phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/294900/294874-59-2.png)
![1H-Benzimidazole, 1-[[2,4,6-tris(1-methylethyl)phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/294900/294874-59-2_b.png)







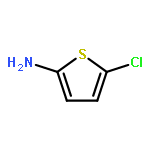
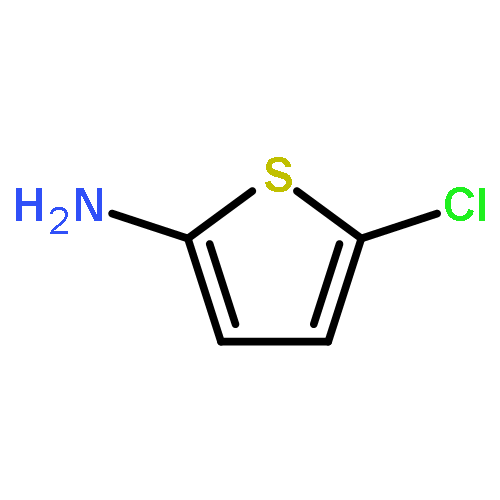




![METHYL 3-[(METHYLSULFONYL)AMINO]BENZOATE](http://img.cochemist.com/ccimg/32100/32087-05-1.png)
![METHYL 3-[(METHYLSULFONYL)AMINO]BENZOATE](http://img.cochemist.com/ccimg/32100/32087-05-1_b.png)

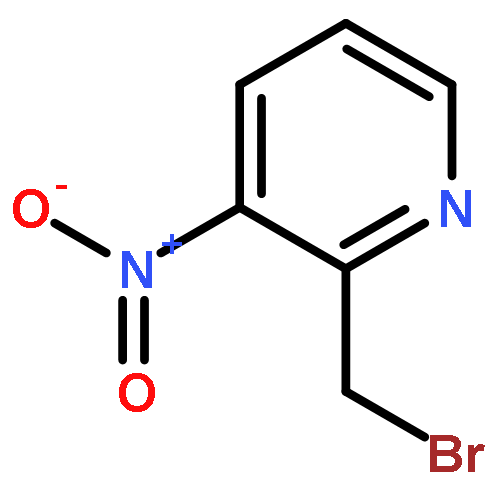
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/17800/17738-71-5.png)
![Acetic acid,2-[(4-chlorophenyl)amino]-2-oxo-](http://img.cochemist.com/ccimg/17800/17738-71-5_b.png)
![5-Chlorobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/13800/13771-75-0.png)
![5-Chlorobenzo[b]thiophene-2-carboxylic acid](http://img.cochemist.com/ccimg/13800/13771-75-0_b.png)
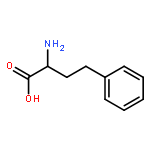
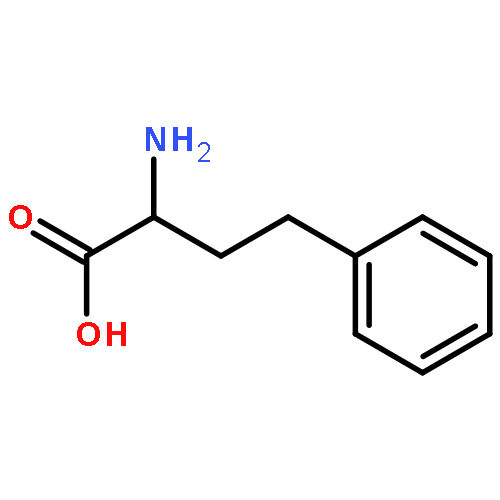
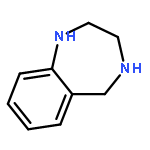
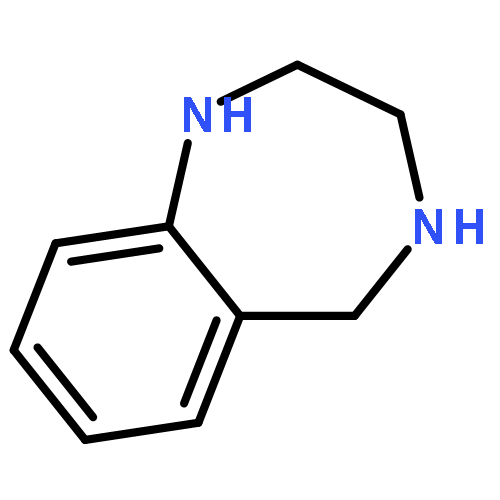


![Acetic acid,2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/4400/4332-50-7.png)
![Acetic acid,2-[(2-chloroethyl)thio]-](http://img.cochemist.com/ccimg/4400/4332-50-7_b.png)
![4,5-DIHYDRO-1H-BENZO[E][1,4]DIAZEPIN-2(3H)-ONE](http://img.cochemist.com/ccimg/1900/1824-72-2.png)
![4,5-DIHYDRO-1H-BENZO[E][1,4]DIAZEPIN-2(3H)-ONE](http://img.cochemist.com/ccimg/1900/1824-72-2_b.png)