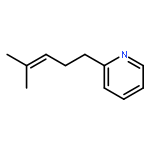
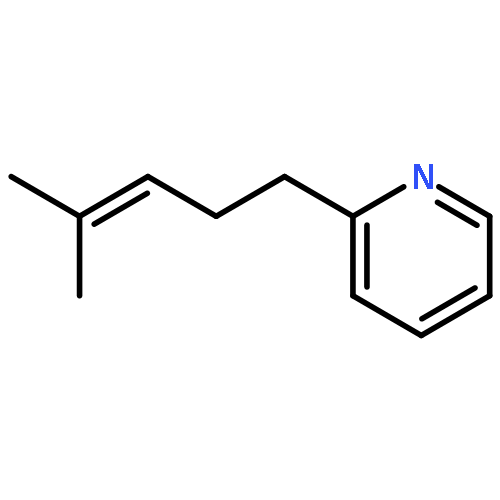
Co-reporter:Donald C. Batesky, Matthew J. Goldfogel, and Daniel J. Weix
The Journal of Organic Chemistry October 6, 2017 Volume 82(Issue 19) pp:9931-9931
Publication Date(Web):September 28, 2017
DOI:10.1021/acs.joc.7b00459
While the use of triphenylphosphine as a reductant is common in organic synthesis, the resulting triphenylphosphine oxide (TPPO) waste can be difficult to separate from the reaction product. While a number of strategies to precipitate TPPO are available, none have been reported to work in more polar solvents. We report here that mixing ZnCl2 with TPPO precipitates a TPPO–Zn complex in high yield in several common polar organic solvents. The solvent compatibility of this procedure and the reliability of the precipitation in the presence of polar functional groups were examined to show the utility and limitations of this method.
Co-reporter:Jill A. Caputo, Leah C. Frenette, Norman Zhao, Kelly L. Sowers, Todd D. Krauss, and Daniel J. Weix
Journal of the American Chemical Society March 29, 2017 Volume 139(Issue 12) pp:4250-4250
Publication Date(Web):March 10, 2017
DOI:10.1021/jacs.6b13379
Photoredox catalysis has become an essential tool in organic synthesis because it enables new routes to important molecules. However, the best available molecular catalysts suffer from high catalyst loadings and rely on precious metals. Here we show that colloidal nanocrystal quantum dots (QDs) can serve as efficient and robust, precious-metal free, photoassisted redox catalysts. A single-sized CdSe quantum dot (3.0 ± 0.2 nm) can replace several different dye catalysts needed for five different photoredox reactions (β-alkylation, β-aminoalkylation, dehalogenation, amine arylation, and decarboxylative radical formation). Even without optimization of the QDs or the reaction conditions, efficiencies rivaling those of the best available metal dyes were obtained.
Co-reporter:Eric C. Hansen, Changfeng Li, Sihang Yang, Dylan Pedro, and Daniel J. Weix
The Journal of Organic Chemistry July 21, 2017 Volume 82(Issue 14) pp:7085-7085
Publication Date(Web):July 6, 2017
DOI:10.1021/acs.joc.7b01334
Despite their importance, the synthesis of alkylated heterocycles from the cross-coupling of Lewis basic nitrogen heteroaryl halides with alkyl halides remains a challenge. We report here a general solution to this challenge enabled by a new collection of ligands based around 2-pyridyl-N-cyanocarboxamidine and 2-pyridylcarboxamidine cores. Both primary and secondary alkyl halides can be coupled with 2-, 3-, and 4-pyridyl halides as well as other more complex heterocycles in generally good yields (41 examples, 69% ave yield).
Co-reporter:Kierra M. M. Huihui, Ruja Shrestha, and Daniel J. Weix
Organic Letters 2017 Volume 19(Issue 2) pp:
Publication Date(Web):January 5, 2017
DOI:10.1021/acs.orglett.6b03509
Conjugate addition of organometallic reagents to enones to form silyl enol ether products is a versatile method to difunctionalize activated olefins, but the organometallic reagents required can be limiting. The reductive cross-electrophile coupling of unhindered primary alkyl bromides with enones and chlorosilanes to form silyl enol ether products is catalyzed by a nickel-complexed ortho-brominated terpyridine ligand. The conditions are compatible with a variety of cyclic/acyclic enones and functional groups.
Co-reporter:Dr. Liangbin Huang;Astrid M. Olivares; Dr. Daniel J. Weix
Angewandte Chemie International Edition 2017 Volume 56(Issue 39) pp:11901-11905
Publication Date(Web):2017/09/18
DOI:10.1002/anie.201706781
AbstractA new method for the synthesis of terminal and internal alkynes from the nickel-catalyzed decarboxylative coupling of N-hydroxyphthalimide esters and bromoalkynes is presented. This reductive cross-electrophile coupling is the first to use a C(sp)−X electrophile, and appears to proceed via an alkynylnickel intermediate. The internal alkyne products are obtained in yields of 41–95 % without the need for a photocatalyst, light, or a strong oxidant. The reaction displays a broad scope of carboxylic acid and alkyne coupling partners, and can tolerate an array of functional groups, including carbamate NH, halogen, nitrile, olefin, ketone, and ester moieties. Mechanistic studies suggest that this process does not involve an alkynylmanganese reagent and instead proceeds through nickel-mediated bond formation.
Co-reporter:Dr. Liangbin Huang;Astrid M. Olivares; Dr. Daniel J. Weix
Angewandte Chemie 2017 Volume 129(Issue 39) pp:12063-12067
Publication Date(Web):2017/09/18
DOI:10.1002/ange.201706781
AbstractA new method for the synthesis of terminal and internal alkynes from the nickel-catalyzed decarboxylative coupling of N-hydroxyphthalimide esters and bromoalkynes is presented. This reductive cross-electrophile coupling is the first to use a C(sp)−X electrophile, and appears to proceed via an alkynylnickel intermediate. The internal alkyne products are obtained in yields of 41–95 % without the need for a photocatalyst, light, or a strong oxidant. The reaction displays a broad scope of carboxylic acid and alkyne coupling partners, and can tolerate an array of functional groups, including carbamate NH, halogen, nitrile, olefin, ketone, and ester moieties. Mechanistic studies suggest that this process does not involve an alkynylmanganese reagent and instead proceeds through nickel-mediated bond formation.
Co-reporter:Kierra M. M. Huihui; Jill A. Caputo; Zulema Melchor; Astrid M. Olivares; Amanda M. Spiewak; Keywan A. Johnson; Tarah A. DiBenedetto; Seoyoung Kim; Laura K. G. Ackerman;Daniel J. Weix
Journal of the American Chemical Society 2016 Volume 138(Issue 15) pp:5016-5019
Publication Date(Web):March 30, 2016
DOI:10.1021/jacs.6b01533
A new method for the decarboxylative coupling of alkyl N-hydroxyphthalimide esters (NHP esters) with aryl iodides is presented. In contrast to previous studies that form alkyl radicals from carboxylic acid derivatives, no photocatalyst, light, or arylmetal reagent is needed, only nickel and a reducing agent (Zn). Methyl, primary, and secondary alkyl groups can all be coupled in good yield (77% ave yield). One coupling with an acid chloride is also presented. Stoichiometric reactions of (dtbbpy)Ni(2-tolyl)I with an NHP ester show for the first time that arylnickel(II) complexes can directly react with NHP esters to form alkylated arenes.
Co-reporter:Liangbin Huang and Daniel J. Weix
Organic Letters 2016 Volume 18(Issue 20) pp:5432-5435
Publication Date(Web):October 13, 2016
DOI:10.1021/acs.orglett.6b02862
Ruthenium ligated to tricyclohexylphosphine or di-tert-butylbipyridine catalyzes the arylation of carboxylic acids with diverse aryl halides (iodide, bromide, and triflate; aryl and heteroaryl). In addition, arylations with 2-iodophenol formed benzochromenones, carboxylate was shown to be a stronger donor than an amide, and the arylation of a pyridine carboxylate was demonstrated. Stoichiometric studies demonstrated that the added ligand is required for reaction with the electrophile but not the C–H bond.
Co-reporter:Dr. Lukiana L. Anka-Lufford;Kierra M. M. Huihui;Dr. Nicholas J. Gower;Dr. Laura K. G. Ackerman ;Dr. Daniel J. Weix
Chemistry - A European Journal 2016 Volume 22( Issue 33) pp:11564-11567
Publication Date(Web):
DOI:10.1002/chem.201602668
Abstract
Cross-electrophile coupling of aryl halides with alkyl halides has thus far been primarily conducted with stoichiometric metallic reductants in amide solvents. This report demonstrates that the use of tetrakis(dimethylamino)ethylene (TDAE) as an organic reductant enables the use of non-amide solvents, such as acetonitrile or propylene oxide, for the coupling of benzyl chlorides and alkyl iodides with aryl halides. Furthermore, these conditions work for several electron-poor heterocycles that are easily reduced by manganese. Finally, we demonstrate that TDAE addition can be used as a control element to ‘hold’ a reaction without diminishing yield or catalyst activity.
Co-reporter:Keywan A. Johnson;Dr. Soumik Biswas ; Daniel J. Weix
Chemistry - A European Journal 2016 Volume 22( Issue 22) pp:7399-7402
Publication Date(Web):
DOI:10.1002/chem.201601320
Abstract
An improved method for the reductive coupling of aryl and vinyl bromides with alkyl halides that gave high yields for a variety of substrates at room temperature with a low (2.5 to 0.5 mol %) catalyst loading is presented. Under the optimized conditions, difficult substrates, such as unhindered alkenyl bromides, can be coupled to give the desired olefins with minimal diene formation and good stereoretention. These improved conditions also worked well for aryl bromides. For example, a gram-scale reaction was demonstrated with 0.5 mol % catalyst loading, whereas reactions at 10 mol % catalyst loading completed in as little as 20 minutes. Finally, a low-cost single-component pre-catalyst, (bpy)NiI2 (bpy=2,2′-bipyridine) that is both air- and moisture-stable over a period of months was introduced.
Co-reporter:Daniel J. Weix
Accounts of Chemical Research 2015 Volume 48(Issue 6) pp:1767
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.accounts.5b00057
Cross-electrophile coupling, the cross-coupling of two different electrophiles, avoids the need for preformed carbon nucleophiles, but development of general methods has lagged behind cross-coupling and C–H functionalization. A central reason for this slow development is the challenge of selectively coupling two substrates that are alike in reactivity. This Account describes the discovery of generally cross-selective reactions of aryl halides and acyl halides with alkyl halides, the mechanistic studies that illuminated the underlying principles of these reactions, and the use of these fundamental principles in the rational design of new cross-electrophile coupling reactions.Although the coupling of two different electrophiles under reducing conditions often leads primarily to symmetric dimers, the subtle differences in reactivity of aryl halides and alkyl halides with nickel catalysts allowed for generally cross-selective coupling reactions. These conditions could also be extended to the coupling of acyl halides with alkyl halides. These reactions are exceptionally functional group tolerant and can be assembled on the benchtop.A combination of stoichiometric and catalytic studies on the mechanism of these reactions revealed an unusual radical-chain mechanism and suggests that selectivity arises from (1) the preference of nickel(0) for oxidative addition to aryl halides and acyl halides over alkyl halides and (2) the greater propensity of alkyl halides to form free radicals. Bipyridine-ligated arylnickel intermediates react with alkyl radicals to efficiently form, after reductive elimination, new C–C bonds. Finally, the resulting nickel(I) species is proposed to regenerate an alkyl radical to carry the chain.Examples of new reactions designed using these principles include carbonylative coupling of aryl halides with alkyl halides to form ketones, arylation of epoxides to form β-aryl alcohols, and coupling of benzyl sulfonate esters with aryl halides to form diarylmethanes. Arylnickel(II) intermediates can insert carbon monoxide to form acylnickel(II) intermediates that react with alkyl halides to form ketones, demonstrating the connection between the mechanisms of reactions of aryl halides and acid chlorides with alkyl halides. The low reactivity of epoxides with nickel can be overcome by the use of either titanium or iodide cocatalysis to facilitate radical generation and this can also be extended to enantioselective arylation of meso-epoxides. The high reactivity of benzyl bromide with nickel, which leads to the formation of bibenzyl in attempted reactions with bromobenzene, can be overcome by using a benzyl mesylate along with cobalt phthalocyanine cocatalysis to convert the mesylate into an alkyl radical.
Co-reporter:Yang Zhao;Daniel J. Weix
Journal of the American Chemical Society 2015 Volume 137(Issue 9) pp:3237-3240
Publication Date(Web):February 25, 2015
DOI:10.1021/jacs.5b01909
The first enantioselective cross-electrophile coupling of aryl bromides with meso-epoxides to form trans-β-arylcycloalkanols is presented. The reaction is catalyzed by a combination of (bpy)NiCl2 and a chiral titanocene under reducing conditions. Yields range from 57 to 99% with 78–95% enantiomeric excess. The 30 examples include a variety of functional groups (ether, ester, ketone, nitrile, ketal, trifluoromethyl, sulfonamide, sulfonate ester), both aryl and vinyl halides, and five- to seven-membered rings. The intermediacy of a carbon radical is strongly suggested by the conversion of cyclooctene monoxide to an aryl [3.3.0]bicyclooctanol.
Co-reporter:Chi Chen; Maxwell B. Hecht; Aydin Kavara; William W. Brennessel; Brandon Q. Mercado; Daniel J. Weix;Patrick L. Holland
Journal of the American Chemical Society 2015 Volume 137(Issue 41) pp:13244-13247
Publication Date(Web):October 7, 2015
DOI:10.1021/jacs.5b08611
Alkene hydrosilylation is typically performed with Pt catalysts, but inexpensive base-metal catalysts would be preferred. We report a Co catalyst for anti-Markovnikov alkene hydrosilylation that can be used without added solvent at low temperatures with low loadings, and can be generated in situ from an air-stable precursor that is simple to synthesize from low-cost, commercially available materials. In addition, a mixture of Co catalysts performs a tandem catalytic alkene isomerization/hydrosilylation reaction that converts multiple isomers of hexene to the same terminal product. This regioconvergent reaction uses isomerization as a benefit rather than a hindrance.
Co-reporter:Laura K. G. Ackerman, Lukiana L. Anka-Lufford, Marina Naodovic and Daniel J. Weix
Chemical Science 2015 vol. 6(Issue 2) pp:1115-1119
Publication Date(Web):10 Nov 2014
DOI:10.1039/C4SC03106G
The nickel-catalyzed cross-coupling of aryl halides with alkyl radicals derived from alkyl halides has recently been extended to couplings with carbon radicals generated by a co-catalyst. In this study, a new co-catalyst, cobalt phthalocyanine (Co(Pc)), is introduced and demonstrated to be effective for coupling substrates not prone to homolysis. This is because Co(Pc) reacts with electrophiles by an SN2 mechanism instead of by the electron-transfer or halogen abstraction mechanisms previously explored. Studies demonstrating the orthogonal reactivity of (bpy)Ni and Co(Pc), applying this selectivity to the coupling of benzyl mesylates with aryl halides, and the adaptation of these conditions to the less reactive benzyl phosphate ester and an enantioconvergent reaction are presented.
Co-reporter:Laura K. G. Ackerman, Lukiana L. Anka-Lufford, Marina Naodovic and Daniel J. Weix
Chemical Science 2015 vol. 6(Issue 6) pp:3633-3633
Publication Date(Web):24 Apr 2015
DOI:10.1039/C5SC90021B
Correction for ‘Cobalt co-catalysis for cross-electrophile coupling: diarylmethanes from benzyl mesylates and aryl halides’ by Laura K. G. Ackerman et al., Chem. Sci., 2015, 6, 1115–1119.
Co-reporter:Stephanie C.M. Dorn, Andrew K Olsen, Rachel E. Kelemen, Ruja Shrestha, Daniel J. Weix
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3365-3367
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2015.02.120
The direct, regioselective, and stereoselective arylation of activated alkynes with aryl iodides using a nickel catalyst and manganese reductant is described. The reaction conditions are mild (40 °C in MeOH, no acid or base) and an intermediate organomanganese reagent is unlikely. Functional groups tolerated include halides and pseudohalides, free and protected anilines, and a benzyl alcohol. Other activated alkynes including an amide and a ketone also reacted to form arylated products in good yields.
Co-reporter:Alexander C. Wotal, Ryan D. Ribson, and Daniel J. Weix
Organometallics 2014 Volume 33(Issue 20) pp:5874-5881
Publication Date(Web):July 10, 2014
DOI:10.1021/om5004682
Acylnickel(II) complexes feature prominently in cross-electrophile coupling (XEC) reactions that form ketones, yet their reactivity has not been systematically investigated. We present here our studies on the reactivity of acylnickel(II) complexes with a series of carbon electrophiles. Bromobenzene, α-chloroethylbenzene, bromooctane, and iodooctane were reacted with (dtbbpy)NiII(C(O)C5H11)(Br) (1b) and (dtbbpy)NiII(C(O)tolyl)(Br) (1c) to form a variety of organic products. While reactions with bromobenzene formed complex mixtures of ketones, reactions with α-chloroethylbenzene were highly selective for the cross-ketone product. Reactions with iodooctane and bromooctane also produced the cross-ketone product, but in intermediate yield and selectivity. In most cases the presence or absence of a chemical reductant (zinc) had only a small effect on the selectivity of the reaction. The coupling of 1c with iodooctane (60% yield) was translated into a catalytic reaction, the carbonylative coupling of bromoarenes with primary bromoalkanes (six examples, 60% average yield).
Co-reporter:Daniel A. Everson and Daniel J. Weix
The Journal of Organic Chemistry 2014 Volume 79(Issue 11) pp:4793-4798
Publication Date(Web):May 13, 2014
DOI:10.1021/jo500507s
A critical overview of the catalytic joining of two different electrophiles, cross-electrophile coupling (XEC), is presented with an emphasis on the central challenge of cross-selectivity. Recent synthetic advances and mechanistic studies have shed light on four possible methods for overcoming this challenge: (1) employing an excess of one reagent; (2) electronic differentiation of starting materials; (3) catalyst–substrate steric matching; and (4) radical chain processes. Each method is described using examples from the recent literature.
Co-reporter:Michael T. Robo, Michael R. Prinsell, and Daniel J. Weix
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:10624-10628
Publication Date(Web):October 24, 2014
DOI:10.1021/jo501925s
Alkylated terpyridine ligands are an increasingly important component of catalysis and dyes but are costly because their synthesis is challenging and often low-yielding. We report an improved method for the Pd/C-catalyzed dehydrogenative coupling of 4-picoline to form the bi- and terpyridine. The addition of MnO2 improves the yield of the reaction, making the reaction useful on a large scale (up to 200 mmol). The use of Pd(OAc)2 or Pd/C/pivalic acid leads to the selective formation of bipyridine.
Co-reporter:Yang Zhao ;Daniel J. Weix
Journal of the American Chemical Society 2013 Volume 136(Issue 1) pp:48-51
Publication Date(Web):December 16, 2013
DOI:10.1021/ja410704d
Epoxides are versatile intermediates in organic synthesis, but have rarely been employed in cross-coupling reactions. We report that bipyridine-ligated nickel can mediate the addition of functionalized aryl halides, a vinyl halide, and a vinyl triflate to epoxides under reducing conditions. For terminal epoxides, the regioselectivity of the reaction depends upon the cocatalyst employed. Iodide cocatalysis results in opening at the less hindered position via an iodohydrin intermediate. Titanocene cocatalysis results in opening at the more hindered position, presumably via TiIII-mediated radical generation. 1,2-Disubstituted epoxides are opened under both conditions to form predominantly the trans product.
Co-reporter:Soumik Biswas ;Daniel J. Weix
Journal of the American Chemical Society 2013 Volume 135(Issue 43) pp:16192-16197
Publication Date(Web):August 18, 2013
DOI:10.1021/ja407589e
The direct cross-coupling of two different electrophiles, such as an aryl halide with an alkyl halide, offers many advantages over conventional cross-coupling methods that require a carbon nucleophile. Despite its promise as a versatile synthetic strategy, a limited understanding of the mechanism and origin of cross selectivity has hindered progress in reaction development and design. Herein, we shed light on the mechanism for the nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides and demonstrate that the selectivity arises from an unusual catalytic cycle that combines both polar and radical steps to form the new C–C bond.
Co-reporter:Ruja Shrestha ; Stephanie C. M. Dorn ;Daniel J. Weix
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:751-762
Publication Date(Web):December 27, 2012
DOI:10.1021/ja309176h
An alternative method to copper-catalyzed conjugate addition followed by enolate silylation for the synthesis of β-disubstituted silyl enol ether products (R1(R2)HCCH═C(OSiR43)R3) is presented. This method uses haloarenes instead of nucleophilic aryl reagents. Nickel ligated to either neocuproine or bipyridine couples an α,β-unsaturated ketone or aldehyde (R2HC═CHC(O)R3) with an organic halide (R1–X) in the presence of a trialkylchlorosilane reagent (Cl–SiR43). Reactions are assembled on the benchtop and tolerate a variety of functional groups (aldehyde, ketone, nitrile, sulfone, pentafluorosulfur, and N-aryltrifluoroacetamide), electron-rich iodoarenes, and electron-poor haloarenes. Mechanistic studies have confirmed the first example of a catalytic reductive conjugate addition of organic halides that proceeds via an allylnickel intermediate. Selectivity is attributed to (1) rapid, selective reaction of LNi0 with chlorotriethylsilane and enone in the presence of other organic electrophiles, and (2) minimization of enone dimerization by ligand steric effects.
Co-reporter:Daniel A. Everson ; Brittany A. Jones ;Daniel J. Weix
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6146-6159
Publication Date(Web):March 30, 2012
DOI:10.1021/ja301769r
A general method is presented for the synthesis of alkylated arenes by the chemoselective combination of two electrophilic carbons. Under the optimized conditions, a variety of aryl and vinyl bromides are reductively coupled with alkyl bromides in high yields. Under similar conditions, activated aryl chlorides can also be coupled with bromoalkanes. The protocols are highly functional-group tolerant (−OH, −NHTs, −OAc, −OTs, −OTf, −COMe, −NHBoc, −NHCbz, −CN, −SO2Me), and the reactions are assembled on the benchtop with no special precautions to exclude air or moisture. The reaction displays different chemoselectivity than conventional cross-coupling reactions, such as the Suzuki–Miyaura, Stille, and Hiyama–Denmark reactions. Substrates bearing both an electrophilic and nucleophilic carbon result in selective coupling at the electrophilic carbon (R–X) and no reaction at the nucleophilic carbon (R–[M]) for organoboron (−Bpin), organotin (−SnMe3), and organosilicon (−SiMe2OH) containing organic halides (X–R–[M]). A Hammett study showed a linear correlation of σ and σ(−) parameters with the relative rate of reaction of substituted aryl bromides with bromoalkanes. The small ρ values for these correlations (1.2–1.7) indicate that oxidative addition of the bromoarene is not the turnover-frequency determining step. The rate of reaction has a positive dependence on the concentration of alkyl bromide and catalyst, no dependence upon the amount of zinc (reducing agent), and an inverse dependence upon aryl halide concentration. These results and studies with an organic reductant (TDAE) argue against the intermediacy of organozinc reagents.
Co-reporter:Lukiana L. Anka-Lufford, Michael R. Prinsell, and Daniel J. Weix
The Journal of Organic Chemistry 2012 Volume 77(Issue 22) pp:9989-10000
Publication Date(Web):October 24, 2012
DOI:10.1021/jo302086g
A general protocol for the coupling of haloarenes with a variety of allylic acetates is presented. Strengths of the method are a tolerance for electrophilic (ketone, aldehyde) and acidic (sulfonamide, trifluoroacetamide) substrates and the ability to couple with a variety of substituted allylic acetates. Secondary alkyl bromides can also be allylated under slightly modified conditions, demonstrating the generality of the approach. Finally, the coupling of a reactive vinyl halide could be achieved by the use of a very hindered ligand and more reactive, branched allylic acetates.
Co-reporter:Ruja Shrestha and Daniel J. Weix
Organic Letters 2011 Volume 13(Issue 10) pp:2766-2769
Publication Date(Web):April 14, 2011
DOI:10.1021/ol200881v
A new method is presented for tandem reductive conjugate addition and silyl enol ether formation from cyclic and acyclic enones and enals in the presence of a Mn reductant, a Ni(terpyridine) catalyst, and a trialkylchlorosilane. The addition of secondary, tertiary, and hindered primary haloalkanes is demonstrated. Preliminary studies on the mechanism show that the intermediacy of L1(Ni)(η3-1-triethylsilyloxyalkenyl)X or in-situ-formed RMnX is unlikely.
Co-reporter:Daniel A. Everson ; Ruja Shrestha ;Daniel J. Weix
Journal of the American Chemical Society 2010 Volume 132(Issue 3) pp:920-921
Publication Date(Web):January 4, 2010
DOI:10.1021/ja9093956
The direct reductive cross-coupling of alkyl halides with aryl halides is described. The transformation is efficient (equimolar amounts of the starting materials are used), generally high-yielding (all but one between 55 and 88% yield), highly functional-group-tolerant [OH, NHBoc, NHCbz, Bpin, C(O)Me, CO2Et, and CN are all tolerated], and easy to perform (uses only benchtop-stable reagents, tolerates small amounts of water and oxygen, changes color when complete, and uses filtration workup). The reaction appears to avoid the formation of intermediate organomanganese species, and a synergistic effect was found when a mixture of two ligands was employed.
Co-reporter:Michael R. Prinsell, Daniel A. Everson and Daniel J. Weix
Chemical Communications 2010 vol. 46(Issue 31) pp:5743-5745
Publication Date(Web):28 Jun 2010
DOI:10.1039/C0CC01716G
The first general method for the reductive dimerization of alkyl halides, alkyl mesylates, alkyl trifluoroacetates, and allylic acetates is reported which proceeds with low catalyst loading (0.5 to 5 mol%), generally high yields (80% ave yield), and good functional-group tolerance.
Co-reporter:Laura K. G. Ackerman, Lukiana L. Anka-Lufford, Marina Naodovic and Daniel J. Weix
Chemical Science (2010-Present) 2015 - vol. 6(Issue 6) pp:NaN3633-3633
Publication Date(Web):2015/04/24
DOI:10.1039/C5SC90021B
Correction for ‘Cobalt co-catalysis for cross-electrophile coupling: diarylmethanes from benzyl mesylates and aryl halides’ by Laura K. G. Ackerman et al., Chem. Sci., 2015, 6, 1115–1119.
Co-reporter:Laura K. G. Ackerman, Lukiana L. Anka-Lufford, Marina Naodovic and Daniel J. Weix
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1119-1119
Publication Date(Web):2014/11/10
DOI:10.1039/C4SC03106G
The nickel-catalyzed cross-coupling of aryl halides with alkyl radicals derived from alkyl halides has recently been extended to couplings with carbon radicals generated by a co-catalyst. In this study, a new co-catalyst, cobalt phthalocyanine (Co(Pc)), is introduced and demonstrated to be effective for coupling substrates not prone to homolysis. This is because Co(Pc) reacts with electrophiles by an SN2 mechanism instead of by the electron-transfer or halogen abstraction mechanisms previously explored. Studies demonstrating the orthogonal reactivity of (bpy)Ni and Co(Pc), applying this selectivity to the coupling of benzyl mesylates with aryl halides, and the adaptation of these conditions to the less reactive benzyl phosphate ester and an enantioconvergent reaction are presented.
Co-reporter:Michael R. Prinsell, Daniel A. Everson and Daniel J. Weix
Chemical Communications 2010 - vol. 46(Issue 31) pp:NaN5745-5745
Publication Date(Web):2010/06/28
DOI:10.1039/C0CC01716G
The first general method for the reductive dimerization of alkyl halides, alkyl mesylates, alkyl trifluoroacetates, and allylic acetates is reported which proceeds with low catalyst loading (0.5 to 5 mol%), generally high yields (80% ave yield), and good functional-group tolerance.