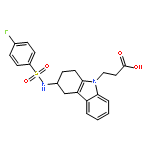
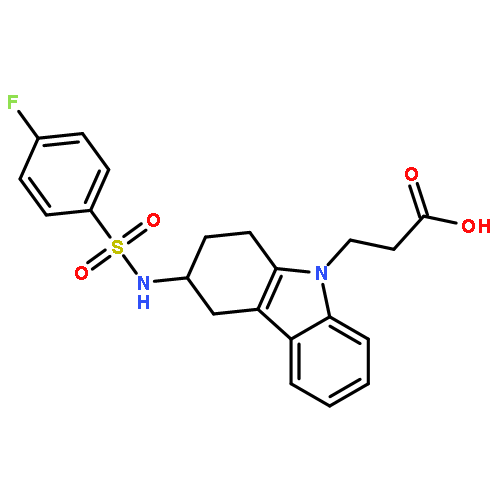
Co-reporter:Mary E. Konkle, Anna L. Blobaum, Christopher W. Moth, Jeffery J. Prusakiewicz, Shu Xu, Kebreab Ghebreselasie, Dapo Akingbade, Aaron T. Jacobs, Carol A. Rouzer, Terry P. Lybrand, and Lawrence J. Marnett
Biochemistry 2016 Volume 55(Issue 2) pp:348-359
Publication Date(Web):December 24, 2015
DOI:10.1021/acs.biochem.5b01222
The cyclooxygenase enzymes (COX-1 and COX-2) are the therapeutic targets of nonsteroidal anti-inflammatory drugs (NSAIDs). Neutralization of the carboxylic acid moiety of the NSAID indomethacin to an ester or amide functionality confers COX-2 selectivity, but the molecular basis for this selectivity has not been completely revealed through mutagenesis studies and/or X-ray crystallographic attempts. We expressed and assayed a number of divergent secondary shell COX-2 active site mutants and found that a COX-2 to COX-1 change at position 472 (Leu in COX-2, Met in COX-1) reduced the potency of enzyme inhibition by a series of COX-2-selective indomethacin amides and esters. In contrast, the potencies of indomethacin, arylacetic acid, propionic acid, and COX-2-selective diarylheterocycle inhibitors were either unaffected or only mildly affected by this mutation. Molecular dynamics simulations revealed identical equilibrium enzyme structures around residue 472; however, calculations indicated that the L472M mutation impacted local low-frequency dynamical COX constriction site motions by stabilizing the active site entrance and slowing constriction site dynamics. Kinetic analysis of inhibitor binding is consistent with the computational findings.
Co-reporter:Loren Baugh, Isolde Le Trong, Patrick S. Stayton, Ronald E. Stenkamp, and Terry P. Lybrand
Biochemistry 2016 Volume 55(Issue 37) pp:5201
Publication Date(Web):September 7, 2016
DOI:10.1021/acs.biochem.6b00698
We report a detailed study of a point mutation of the crucial binding site residue, D128, in the biotin–streptavidin complex. The conservative substitution, D128N, preserves the detailed structure observed for the wild-type complex but has an only minimal impact on biotin binding, even though previous experimental and computational studies suggested that a charged D128 residue was crucial for high-affinity binding. These results show clearly that the fundamental basis for streptavidin’s extremely strong biotin binding affinity is more complex than assumed and illustrate some of the challenges that may arise when analyzing extremely strong ligand–protein binding interactions.
Co-reporter:Sarah J. Edwards, Christopher W. Moth, Sunghoon Kim, Suzanne Brandon, Zheng Zhou, Charles E. Cobb, Eric J. Hustedt, Albert H. Beth, Jarrod A. Smith, and Terry P. Lybrand
The Journal of Physical Chemistry B 2014 Volume 118(Issue 18) pp:4717-4726
Publication Date(Web):April 11, 2014
DOI:10.1021/jp4099705
We report here specialized functions incorporated recently in the rigid-body docking software toolkit TagDock to utilize electron paramagnetic resonance derived (EPR-derived) interresidue distance measurements and spin-label accessibility data. The TagDock package extensions include a custom methanethiosulfonate spin label rotamer library to enable explicit, all-atom spin-label side-chain modeling and scripts to evaluate spin-label surface accessibility. These software enhancements enable us to better utilize the biophysical data routinely available from various spin-labeling experiments. To illustrate the power and utility of these tools, we report the refinement of an ankyrin:CDB3 complex model that exhibits much improved agreement with the EPR distance measurements, compared to model structures published previously.
Co-reporter:Sarah M. Stow, Cody R. Goodwin, Michal Kliman, Brian O. Bachmann, John A. McLean, and Terry P. Lybrand
The Journal of Physical Chemistry B 2014 Volume 118(Issue 48) pp:13812-13820
Publication Date(Web):October 31, 2014
DOI:10.1021/jp509398e
Ion mobility-mass spectrometry (IM-MS) allows the separation of ionized molecules based on their charge-to-surface area (IM) and mass-to-charge ratio (MS), respectively. The IM drift time data that is obtained is used to calculate the ion-neutral collision cross section (CCS) of the ionized molecule with the neutral drift gas, which is directly related to the ion conformation and hence molecular size and shape. Studying the conformational landscape of these ionized molecules computationally provides interpretation to delineate the potential structures that these CCS values could represent, or conversely, structural motifs not consistent with the IM data. A challenge in the IM-MS community is the ability to rapidly compute conformations to interpret natural product data, a class of molecules exhibiting a broad range of biological activity. The diversity of biological activity is, in part, related to the unique structural characteristics often observed for natural products. Contemporary approaches to structurally interpret IM-MS data for peptides and proteins typically utilize molecular dynamics (MD) simulations to sample conformational space. However, MD calculations are computationally expensive, they require a force field that accurately describes the molecule of interest, and there is no simple metric that indicates when sufficient conformational sampling has been achieved. Distance geometry is a computationally inexpensive approach that creates conformations based on sampling different pairwise distances between the atoms within the molecule and therefore does not require a force field. Progressively larger distance bounds can be used in distance geometry calculations, providing in principle a strategy to assess when all plausible conformations have been sampled. Our results suggest that distance geometry is a computationally efficient and potentially superior strategy for conformational analysis of natural products to interpret gas-phase CCS data.
Co-reporter:Jarrod A. Smith, Sarah J. Edwards, Christopher W. Moth, and Terry P. Lybrand
Biochemistry 2013 Volume 52(Issue 33) pp:
Publication Date(Web):July 22, 2013
DOI:10.1021/bi400158k
We report here new computational tools and strategies to efficiently generate three-dimensional models for oligomeric biomolecular complexes in cases where there is limited experimental restraint data to guide the docking calculations. Our computational tools are designed to rapidly and exhaustively enumerate all geometrically possible docking poses for an oligomeric complex, rather than generate detailed, atomic-resolution models. Experimental data, such as interatomic distance measurements, are then used to select and refine docking poses that are consistent with the experimental restraints. Our computational toolkit is designed for use with sparse data sets to generate intermediate-resolution docking models, and utilizes distance difference matrix analysis to identify further restraint measurements that will provide maximum additional structural refinement. Thus, these tools can be used to help plan optimal residue positions for probe incorporation in labor-intensive biophysical experiments such as chemical cross-linking, electron paramagnetic resonance, or Förster resonance energy transfer spectroscopy studies. We present benchmark results for docking the collection of all 176 heterodimer protein complexes from the ZDOCK database, as well as a protein homodimer with recently collected experimental distance restraints, to illustrate the toolkit’s capabilities and performance, and to demonstrate how distance difference matrix analysis can automatically identify and prioritize additional restraint measurements that allow us to rapidly optimize docking poses.
Co-reporter:Loren Baugh, Isolde Le Trong, David S. Cerutti, Nital Mehta, Susanne Gülich, Patrick S. Stayton, Ronald E. Stenkamp, and Terry P. Lybrand
Biochemistry 2012 Volume 51(Issue 2) pp:
Publication Date(Web):December 6, 2011
DOI:10.1021/bi201221j
We report a point mutation in the second contact shell of the high-affinity streptavidin–biotin complex that appears to reduce binding affinity through transmitted effects on equilibrium dynamics. The Y54F streptavidin mutation causes a 75-fold loss of binding affinity with 73-fold faster dissociation, a large loss of binding enthalpy (ΔΔH = 3.4 kcal/mol at 37 °C), and a small gain in binding entropy (TΔΔS = 0.7 kcal/mol). The removed Y54 hydroxyl is replaced by a water molecule in the bound structure, but there are no observable changes in structure in the first contact shell and no additional changes surrounding the mutation. Molecular dynamics simulations reveal a large increase in the atomic fluctuation amplitudes for W79, a key biotin contact residue, compared to the fluctuation amplitudes in the wild-type. The increased W79 atomic fluctuation amplitudes are caused by loss of water-mediated hydrogen bonds between the Y54 hydroxyl group and peptide backbone atoms in and near W79. We propose that the increased atomic fluctuation amplitudes diminish the integrity of the W79–biotin interaction and represents a loosening of the “tryptophan collar” that is critical to the slow dissociation and high affinity of streptavidin–biotin binding. These results illustrate how changes in protein dynamics distal to the ligand binding pocket can have a profound impact on ligand binding, even when equilibrium structure is unperturbed.
Co-reporter:Loren Baugh, Isolde Le Trong, David S. Cerutti, Susanne Gülich, Patrick S. Stayton, Ronald E. Stenkamp and Terry P. Lybrand
Biochemistry 2010 Volume 49(Issue 22) pp:
Publication Date(Web):May 12, 2010
DOI:10.1021/bi1005392
We have identified a distal point mutation in streptavidin that causes a 1000-fold reduction in biotin binding affinity without disrupting the equilibrium complex structure. The F130L mutation creates a small cavity occupied by a water molecule; however, all neighboring side chain positions are preserved, and protein−biotin hydrogen bonds are unperturbed. Molecular dynamics simulations reveal a reduced mobility of biotin binding residues but no observable destabilization of protein−ligand interactions. Our combined structural and computational studies suggest that the additional water molecule may affect binding affinity through an electronic polarization effect that impacts the highly cooperative hydrogen bonding network in the biotin binding pocket.
Co-reporter:David S. Cerutti, Isolde Le Trong, Ronald E. Stenkamp and Terry P. Lybrand
The Journal of Physical Chemistry B 2009 Volume 113(Issue 19) pp:6971-6985
Publication Date(Web):April 17, 2009
DOI:10.1021/jp9010372
We present a 250 ns simulation of the wild-type, biotin-liganded streptavidin tetramer in the solution phase and compare the trajectory to two previously published simulations of the protein in its crystal lattice. By performing both types of simulations, we are able to interpret the protein’s behavior in solution in the context of its X-ray structure. We find that the rate of conformational sampling is increased in solution over the lattice environment, although the relevant conformational space in solution is also much larger, as indicated by overall fluctuations in the positions of backbone atoms. We also compare the distributions of χ1 angles sampled by side chains exposed to solvent in the lattice and in the solution phase, obtaining overall good agreement between the distributions obtained in our most rigorous lattice simulation and the crystallographic χ1 angles. We observe changes in the χ1 distributions in the solution phase, and note an apparent progression of the distributions as the environment changes from a tightly packed lattice filled with crystallization media to a bath of pure water. Finally, we examine the interaction of biotin and streptavidin in each simulation, uncovering a possible alternate conformation of the biotin carboxylate tail. We also note that a hydrogen bond observed to break transiently in previous solution-phase simulations is predominantly broken in this much longer solution-phase trajectory; in the lattice simulations, the lattice environment appears to help maintain the hydrogen bond, but more sampling will be needed to confirm whether the simulation model truly gives good agreement with the X-ray data in the lattice simulations. We expect that pairing solution-phase biomolecular simulations with crystal lattice simulations will help to validate simulation models and improve the interpretation of experimentally determined structures.
Co-reporter:David S. Cerutti, Isolde Le Trong, Ronald E. Stenkamp and Terry P. Lybrand
Biochemistry 2008 Volume 47(Issue 46) pp:
Publication Date(Web):October 24, 2008
DOI:10.1021/bi800894u
A 250 ns molecular dynamics simulation of the biotin-liganded streptavidin crystal lattice, including cryoprotectant molecules and crystallization salts, is compared to a 250 ns simulation of the lattice solvated with pure water. The simulation using detailed crystallization conditions preserves the initial X-ray structure better than the simulation using pure water, even though the protein molecules display comparable mobility in either simulation. Atomic fluctuations computed from the simulation with crystallization conditions closely reproduce fluctuations derived from experimental temperature factors (correlation coefficient of 0.88, omitting two N-terminal residues with very high experimental B-factors). In contrast, fluctuations calculated from the simulation with pure water were less accurate, particularly for two of the streptavidin loops exposed to solvent in the crystal lattice. Finally, we obtain good agreement between the water and cryoprotectant densities obtained from the simulated crystallization conditions and the electron density due to solvent molecules in the X-ray structure. Our results suggest that detailed lattice simulations with realistic crystallization conditions can be used to assess potential function parameters, validate simulation protocols, and obtain valuable insights that solution-phase simulations do not easily provide. We anticipate that this will prove to be a powerful strategy for molecular dynamics simulations of biomolecules.
Co-reporter:Eric S. Dawson;Rel M. Henne;Laurence J. Miller;Terry P. Lybr
Basic & Clinical Pharmacology & Toxicology 2002 Volume 91(Issue 6) pp:
Publication Date(Web):28 JAN 2003
DOI:10.1034/j.1600-0773.2002.910605.x
Abstract: Numerous techniques have been used to elucidate the structural basis for interaction of cholecystokinin (CCK)-related peptides with their hormone-binding receptor, the CCK-A receptor (CCK-AR), including structure-activity relationship studies, site-directed mutagenesis, photoaffinity-labeling, and solution NMR analysis of both CCK peptide ligands and peptide fragments derived from the CCK-A receptor. Different structural models have been developed for the peptide-receptor complexes using various subsets of the available experimental data (Giragossian & Mierke 2001; Ding et al. 2002; Escrieut et al. 2002). Here, we review details of the various models and evaluate the impact of selected experimental data sets on model development.
.jpg)
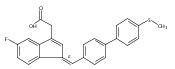
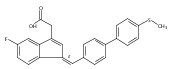


![9H-Fluoren-9-one,2-amino-5-[(2S)-2-hydroxy-3-[(1-methylethyl)amino]propoxy]-](http://img.cochemist.com/ccimg/630500/630400-37-2.png)
![9H-Fluoren-9-one,2-amino-5-[(2S)-2-hydroxy-3-[(1-methylethyl)amino]propoxy]-](http://img.cochemist.com/ccimg/630500/630400-37-2_b.png)

![Acetic acid,2-[[3-[(2R)-2-[[(2R)-2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1H-indol-7-yl]oxy]-](http://img.cochemist.com/ccimg/244100/244081-42-3.png)
![Acetic acid,2-[[3-[(2R)-2-[[(2R)-2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1H-indol-7-yl]oxy]-](http://img.cochemist.com/ccimg/244100/244081-42-3_b.png)
![1H-Indole-3-acetic acid,1-[(4-bromophenyl)methyl]-5-methoxy-2-methyl-](http://img.cochemist.com/ccimg/176200/176174-97-3.png)
![1H-Indole-3-acetic acid,1-[(4-bromophenyl)methyl]-5-methoxy-2-methyl-](http://img.cochemist.com/ccimg/176200/176174-97-3_b.png)
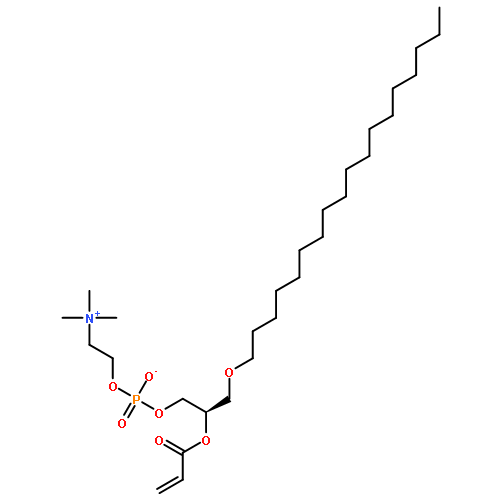
![2-{[(dodecyloxy)(hydroxy)phosphoryl]oxy}-N,N,N-trimethylethanaminium chloride](http://img.cochemist.com/ccimg/54000/53949-18-1.png)
![2-{[(dodecyloxy)(hydroxy)phosphoryl]oxy}-N,N,N-trimethylethanaminium chloride](http://img.cochemist.com/ccimg/54000/53949-18-1_b.png)
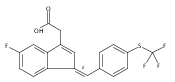
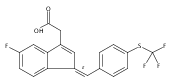
![1H-Indene-3-aceticacid, 5-fluoro-2-methyl-1-[[4-(methylthio)phenyl]methylene]-, (1Z)-](http://img.cochemist.com/ccimg/49700/49627-27-2.png)
![1H-Indene-3-aceticacid, 5-fluoro-2-methyl-1-[[4-(methylthio)phenyl]methylene]-, (1Z)-](http://img.cochemist.com/ccimg/49700/49627-27-2_b.png)





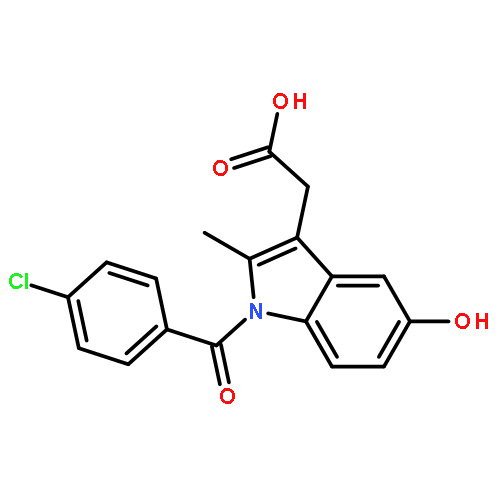
![ethyl [1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl]acetate](http://img.cochemist.com/ccimg/16500/16401-99-3.png)
![ethyl [1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl]acetate](http://img.cochemist.com/ccimg/16500/16401-99-3_b.png)






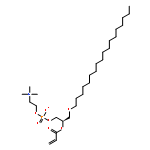


![2-[1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl]acetamide](http://img.cochemist.com/ccimg/6300/6264-33-1.png)
![2-[1-(4-chlorobenzoyl)-5-methoxy-2-methyl-1H-indol-3-yl]acetamide](http://img.cochemist.com/ccimg/6300/6264-33-1_b.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-, (2S)-](/data/chemimg/55200/4199-09-1.png)
![2-Propanol, 1-[(1-methylethyl)amino]-3-(1-naphthalenyloxy)-, (2S)-](/data/chemimg/55200/4199-09-1_b.png)




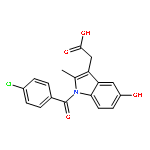











![1,2-Benzenediol,4-[(1R)-1-hydroxy-2-[(1-methylethyl)amino]ethyl]-](http://img.cochemist.com/ccimg/100/51-31-0.png)
![1,2-Benzenediol,4-[(1R)-1-hydroxy-2-[(1-methylethyl)amino]ethyl]-](http://img.cochemist.com/ccimg/100/51-31-0_b.png)