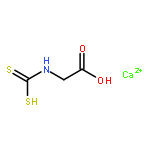
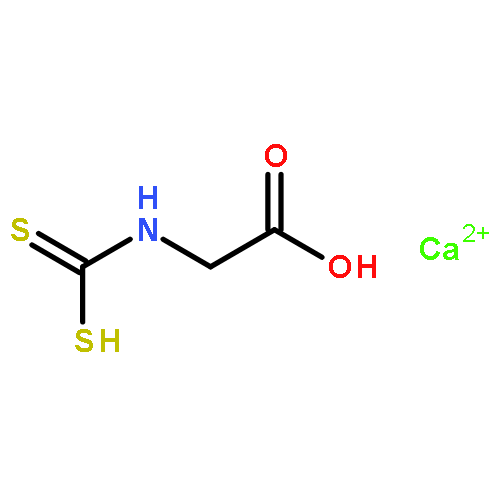
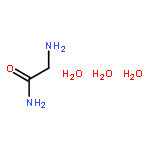
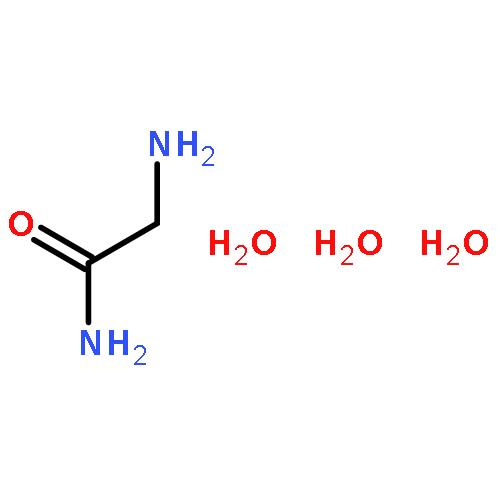
Co-reporter:Zhi Long, Liqin Gao, Yankai Li, Baotao Kang, Jin Yong Lee, Junjie Ge, Changpeng Liu, Shuhua Ma, Zhao Jin, and Hongqi Ai
ACS Applied Materials & Interfaces November 8, 2017 Volume 9(Issue 44) pp:38165-38165
Publication Date(Web):October 25, 2017
DOI:10.1021/acsami.7b11852
The self-assembly powder (SAP) with varying Nafion content was synthesized and characterized by XRD, XPS, HRTEM, and mapping. It is observed that the oxygen from oxygen functional groups transfers to the surface of Pt and generate PtO during the process of self-assembly with the mechanism of micro galvanic cell, where Pt, carbon black, and Nafion act as the anode, cathode and electrolyte, respectively. The appearance of PtO on the surface of Pt leads to a turnover of Nafion structure, and therefore more hydrophilic sulfonic groups directly contact with Pt, and thus the triple-phase boundary (TPB) has been expanded.Keywords: catalyst layers; Nafion; PEMFCs; Pt/C; triple-phase boundary;
Co-reporter:Mingyan Dong, Wei Zhao, Dingkun Hu, Hongqi Ai, and Baotao Kang
ACS Chemical Neuroscience July 19, 2017 Volume 8(Issue 7) pp:1577-1577
Publication Date(Web):April 13, 2017
DOI:10.1021/acschemneuro.7b00080
Amyloid-β (Aβ40/Aβ42) peptide with a length of 40 or 42 residues is naturally secreted as cleavage product of the amyloid precursor protein, and formation of Aβ aggregates in a patient’s brain is a hallmark of Alzheimer’s disease (AD). Therefore, disaggregation and disruption provide potential therapeutic approaches to reduce, inhibit, and even reverse Aβ aggregation. The disaggregation/inhibition effect of the inhibitors applies generally to both Aβ40 and Aβ42 aggregations. Here we capture the atomic-level details of the interaction between Aβ40/Aβ42 and either natural tanshinone compound TS1 or its derivative TS0, and observe novel results by using molecular dynamics simulations. We observe that the natural TS1 indeed inhibits the monomolecular Aβ42 (mAβ42) aggregation and disaggregates Aβ42 amyloid fibrils, being in good agreement with the experimental results. TS1 is favorable to stabilize mAβ40 and even Aβ40 fibril, playing an opposite role to that in the Aβ42 counterpart, however. TS0 can inhibit the misfolding of either mAβ40 or mAβ42 and disaggregate Aβ42 fibril but stabilize the Aβ40 fibril. Using a combination of secondary structural analysis, MM-PBSA binding energy calculations, and radial distribution functions computations, we find that both TS0 and TS1, especially the former, prefer to bind at the charged residues within disordered N-terminus with a scarce positive binding energy and disappear the characteristic C-terminal bend region of Aβ42 fibril, as well as twist the Aβ42 fibril seriously. It turns out to destabilize the Aβ42 fibril and enable the conversion of U-shaped Aβ42 fibril from the onefold to the twofold morphologies. The N-terminal binding preference helps us to identify N-terminal region as the specific epitope for specific inhibitors/drugs (such as TS0 and analogues), heralding unusual inhibition/disaggregation or stabilization mechanisms, and offering an alternative direction in engineering new inhibitors to treat AD.Keywords: Alzheimer’s disease; Aβ; disaggregation/inhibition mechanism; preferential binding at N-terminus region; tanshinone;
Co-reporter:Baotao Kang, Hu Shi, Si Wu, Wei Zhao, Hongqi Ai, Jin Yong Lee
Carbon 2017 Volume 123(Volume 123) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.carbon.2017.07.087
In the present paper, density functional theory calculations were carried out on a detailed oxygen reduction reaction (ORR) process catalysed by nitrogen-doped β-graphyne (βGy). Nitrogen-doping can increase the positive charge of the adjacent carbon atom and the graphyne work function, promoting O2 decomposition. Our calculations revealed that N-doped βGy can efficiently facilitate the ORR process via a four-electron mechanism, regardless of doping position. Additionally, our results revealed that the N doping site was essential for tuning the ORR catalytic activity. N1 doping was shown to more effectively promote the ORR than N2 doping. In addition, our research enhances the current understanding of catalytic activity at the molecular level.Download high-res image (116KB)Download full-size image
Co-reporter:Mingyan Dong, Haoyue Li, Dingkun Hu, Wei Zhao, Xueying Zhu, and Hongqi Ai
ACS Chemical Neuroscience 2016 Volume 7(Issue 5) pp:599
Publication Date(Web):February 12, 2016
DOI:10.1021/acschemneuro.5b00343
The aggregation of amyloid-β (Aβ) peptide induced by Cu2+ is a key factor in development of Alzheimer’s disease (AD), and metal ion chelation therapy enables treatment of AD. Three CQi (i = 1, 2, and 3 with R = H, Cl, and NO2, respectively) drugs had been verified experimentally to be much stronger inhibitors than the pioneer clioquinol (CQ) in both disaggregation of Aβ40 aggregate and reduction of toxicity induced by Cu2+ binding at low pH. Due to the multiple morphologies of Cu2+–Aβ40 complexes produced at different pH states, we performed a series of molecular dynamics simulations to explain the structural changes and morphology characteristics as well as intrinsic disaggregation mechanisms of three Cu2+–Aβ40 models in the presence of any of the three CQi drugs at both low and high pH states. Three inhibition mechanisms for CQi were proposed as “insertion”, “semi-insertion”, and “surface” mechanisms, based on the morphologies of CQi–model x (CQi–x, x = 1, 2, and 3) and the strengths of binding between CQi and the corresponding model x. The insertion mechanism was characterized by the morphology with binding strength of more than 100 kJ/mol and by CQi being inserted or embedded into the hydrophobic cavity of model x. In those CQi–x morphologies with lower binding strength, CQi only attaches on the surface or inserts partly into Aβ peptide. Given the evidence that the binding strength is correlated positively with the effectiveness of drug to inhibit Aβ aggregation and thus to reduce toxicity, the data of binding strength presented here can provide a reference for one to screen drugs. From the point of view of binding strength, CQ2 is the best drug. Because of the special role of Asp23 in both Aβ aggregation and stabilizing the Aβ fibril, the generation of a H-bond between CQ3 and Asp23 of the Aβ40 peptide is believed to be responsible for CQ3 having the strongest disaggregation capacity. Therefore, besides strong binding, stronger propensity to H-bond with Asp23 would be another key factor to be taken seriously into account in drug screens. Meanwhile, the structural characteristics of drug CQi itself are also worthy of attention. First, the increasing polarity from CQ1 and CQ2 to CQ3 in turn results in increasing probability and strength of the interaction between the drug and the N-terminal (NT) region of Aβ40, which obviously inhibits Aβ peptide aggregation induced by Cu2+ binding. Second, both the benzothiazole ring and phenol ring of CQi can overcome the activation energy barrier (∼16 kJ/mol) to rotate flexibly around the intramolecular C7–N14 bond to achieve the maximum match and interaction with the ambient Aβ40 residues. Such a structural feature of CQi paves the new way for ones in selection and modification of a drug.Keywords: CQi inhibitor; Cu2+−Aβ40 aggregation; helix−coil/turn transition; insertion mechanism; rotation bond
Co-reporter:Ran Zhang;Dr. Hongqi Ai;Dr. Xueying Zhu; Qiang Li
ChemPhysChem 2016 Volume 17( Issue 11) pp:1656-1668
Publication Date(Web):
DOI:10.1002/cphc.201600004
Abstract
As the main sequence responsible for metal ion coordination in the amyloid beta (Aβ) peptide, Aβ1–16 plays a key role in the understanding of the aggregation of Aβ induced by Cu2+ ions. There is no consensus on the nature of the coordination sphere of the Cu2+–Aβ complex so far due to the amorphous conformation of the Aβ1–16 peptide itself and the pH dependence of Cu2+–Aβ coordination. The simulation reported here reveals that human Aβ1–16 monomer has a U-shape morphology, which is preserved at any pH. This morphology accommodates Cu2+ ions with several binding sites and is also the basis for establishing a center-distance statistical method (CDSM). Based on this CDSM, specific histidine residues for a Cu2+-coordinated sphere are identified and the corresponding accurate pH range is established, indicating that the CDSM can be used as a reference to predict the potential coordination sites of metal ions in other amorphous peptides. By contrast, mouse Aβ1–16 monomer has a more open and random morphology than human Aβ1–16 due to the differences of three sequence positions. These mutations not only reduce the number of binding sites required by a stable Cu2+-binding sphere but also diminish the capacity to generate salt bridges compared to the human peptide. These observations offer insights into the roles of three residues that differ in the mouse Aβ1–16 and perhaps into the reasons mice seldom develop Alzheimer's disease.
Co-reporter:Mingyan Dong;Thomas J. Paul;Zachary Hoffmann;Dr. Kwaichow Chan;Dingkun Hu;Dr. Hongqi Ai;Dr. Rajeev Prabhakar
ChemPhysChem 2016 Volume 17( Issue 16) pp:2558-2566
Publication Date(Web):
DOI:10.1002/cphc.201600256
Abstract
In this study, structural and mechanical properties of a series of models of Aβ42 (one- and two-fold) and Aβ40 (two- and three-fold) fibrils have been computed by using all-atom molecular dynamics simulations. Based on calculations of the twist angle (θ) and periodicity (v=360d/θ), oligomers formed by 20, 11, and 13 monomers were found to be the smallest realistic models of three-fold Aβ40, one-fold Aβ42, and two-fold Aβ42 fibrils, respectively. Our results predict that the Aβ40 fibrils initially exist in two staggered conformations [STAG(+2) and STAG(+1)] and then undergo a [STAG(+2)STAG(+1)] transformation in a size-dependent manner. The length of the loop region consisting of the residues 23–29 shrinks with the elongation of both Aβ40 and Aβ42 fibrils. A comparison of the computed potential energy suggests that a two-fold Aβ40 aggregate is more stable than its three-fold counterpart, and that Aβ42 oligomers can exist only in one-fold conformation for aggregates of more than 11 monomers in length. The computed Young′s modulus and yield strengths of 50 GPa and 0.95 GPa, respectively, show that these aggregates possess excellent material properties.
Co-reporter:Xueying Zhu
Journal of Molecular Modeling 2016 Volume 22( Issue 7) pp:
Publication Date(Web):2016 July
DOI:10.1007/s00894-016-3009-x
This report presents a systematic investigation of the interactions of water molecule(s) with a series of amino acid cations (Gly+, Ala+, Val+, and Leu+), halogen anions (Cl−, Br−, BF4−, and PF6−), and clusters (GlyCl)n (n = 1–5). The results reveal that H-bonds between amino acid ionic liquids (AAILs) and water molecules are crucial to the properties of aqueous solution of AAILs. The properties of AAIL in water solution depend on the alkyl chain of the amino acid cation, the size of the halogen anion, and the number of water molecules, which provides a certain theoretical basis for the design and application of new AAILs. A series of calculations for some different models showed that quadruple-GlyCl hydrate represents a basic unit for the Gly–water binary system, and can be employed as the simplest model for studying an AAIL–water cluster. On the basis of this model, the effects of water on the hygroscopicity, speed of solubility, viscosity, density, solution enthalpy, and polarity of the AAIL were also predicted. Most importantly, unlike traditional ILs, the novel GlyCl-type AAIL favors interaction of its cationic part, rather than its anionic part, with surrounding water molecules, thus amino acid cationic ILs expand the types of IL available, increasing the choice of ILs for different purposes. We hope that the application of this AAIL in many fields will lead to optimization of this class of compound and be of benefit to the environment.
Co-reporter:Jinpeng Chen;Yongping Zhao ;Jingjing Liu
Journal of Physical Organic Chemistry 2012 Volume 25( Issue 2) pp:126-131
Publication Date(Web):
DOI:10.1002/poc.1881
Stability orderings of 150 stable complexes formed by metal ions (Na+, K+, Ca2+, Mg2+, and Zn2+) and 13 stable thymine tautomers in both solvent and gas phases are obtained, and the optimal binding site for a metal ion in a specific thymine tautomer is identified. Results indicate that the complex with the canonical thymine tautomer (T1) is more stable than those with the rare ones, and the monodentate complex M–T1o4(o2) are their ground-state form in the solvent phase. The ground-state thymine complexes bound by Ca2+, Mg2+, or Zn2+ become bidentate M–T3o4lo2,n3, which is derived from a rare thymine tautomer T3o4l, whereas those bound by Na+ and K+ are still monodentate complexes M–T1o4(o2), however, in the gas phase. The differences in stability are discussed in detail from the binding strength of metal ions, relative energy of the corresponding thymine tautomers, and solution effect. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Hongqi Ai, Jinpeng Chen, and Chong Zhang
The Journal of Physical Chemistry B 2012 Volume 116(Issue 46) pp:13624-13636
Publication Date(Web):October 30, 2012
DOI:10.1021/jp308937k
Tautomerization processes of amino–imino adenine isomer (A → A1) in five different environments are studied by the density functional theory (B3LYP) method. The five environments are metal ion (M, M = K+, Na+, Cu+, Zn+, Ca2+, Mg2+, Cu2+, Zn2+) coordinated bidentate system, either monowater (W) or monoammonia (N) attached system, both metal ion and monowater cooperative system (M-W), and both metal ion and monoammonia cooperative system (M-N). Results show that the complexes formed by noncanonical rare imino form A1 are more stable than those formed by the canonical amino one in most of these environments. The tautomerization of A → A1 becomes quite easy in either M-W or M-N system. It is noteworthy that under divalent M-N environment the A → A1 process meets with particularly lower and even free energy barrier, indicating the instability of the amino adenine isomer and probable existence of more stable imino adenine isomer. Expanding studies for the microhydration at the metal ion of the M-N system predict the required number (n) of water molecules to remain the amino adenine isomer A (AMNnW) stable. The number of n is 2, 3, 3, and 4 for M = Ca2+, Zn2+, Cu2+, and Mg2+, respectively. The present study provides further understanding for the amino–imino tautomerization behavior of the most stable adenine under the influence of several related closely factors, and is useful for rational design of these different environments for the purposes of prevention and control of pyrimidines mispairing, which is responsible for the mutagenic properties of the nucleic acid bases.
Co-reporter:Hongqi Ai;Chong Zhang;Wei He;Kwaichow Chan;Qiang Li
Journal of Molecular Modeling 2012 Volume 18( Issue 1) pp:53-64
Publication Date(Web):2012 January
DOI:10.1007/s00894-011-1026-3
In the present paper we employ a series of dicationized (K+& C+; C = H, Li, Na, or K) hexapeptide (G6C+K+x, x = 1, 2, … or 5) complexes in gas phase (GP) and aqueous phase (AP) as models to mimic the key characteristics of the real biosystems. An interesting phenomenon is observed that the binding properties of K+x to G6C+ species change in the two phases, i.e., those positive binding energies in the GP become negative ones in the AP. Then we probe the origin of property change of the binding energies in the two different phases and associate these changes with some biological phenomena.
Co-reporter:Dejie Li and Hongqi Ai
The Journal of Physical Chemistry B 2009 Volume 113(Issue 34) pp:11732-11742
Publication Date(Web):July 31, 2009
DOI:10.1021/jp9031833
In this work, the three most stable uracil isomers (U1, U2, and U3) and their neutral, positive, and negative charged multihydrates are chosen as research objects to investigate the tautomeric process between the most stable uracil, U1, and its two minor stable isomers, U2 and U3. By the study, deeper insight can be obtained regarding point mutations induced by uracil deformation. Toward the target, the activation energies of the intramolecular proton transfer (tautomeric process) as well as the catalysis effects of water molecules and of charges attached are investigated using density functional theory (DFT) calculations by means of the B3LYP exchange and correlation functions. Results reveal that water molecules hold a stronger catalysis effect on the proton transfer in these negative charged uracil hydrates than in the neutral counterparts. The optimal number of water molecules needed to catalyze the proton transfer is determined as two in the neutral hydrated systems, whereas it is three in the negative charged systems. Positive charge attachment, however, hinders the intramolecualr proton transfer of uracil, and the charge and the proton of uracil will transfer to the water clusters if water molecules are attached. Then the positive charged hydrates look more like U1a/b+[(H2O)n+H+] species in structure. Analysis reveals that it is the acceptance process of the last proton to determine the impossibility of proton transfer and result in the failure of tautomeric processes from cat-U1a-nw to cat-U2-nw and from cat-U1b-nw to cat-U3-nw. Detailed structural parameters and energy changes are discussed for the above different processes.
Co-reporter:Hongqi Ai, Yun Li, Chong Zhang, Jijun Feng
Chemical Physics 2007 Volume 334(1–3) pp:64-76
Publication Date(Web):20 April 2007
DOI:10.1016/j.chemphys.2007.02.015
Abstract
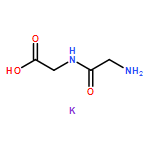
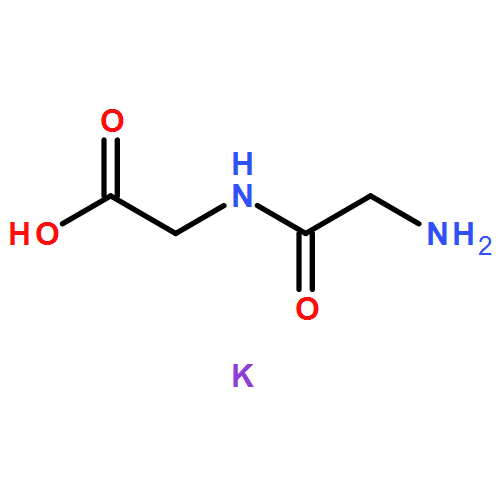
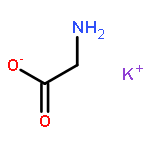
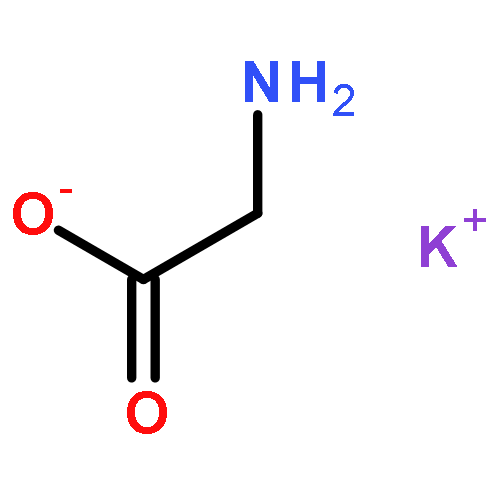
The dependence of binding energy (BE) on the binding sites of both protonated and various metalated (M+/2+ = Li+, Na+ or K+, and Be2+, Mg2+ or Ca2+) oligoglycine derivatives, GnH+Mx+/2+ (G = glycine and H+ = proton; n = 2, 3 and x = 1–3) in the gas phase has been determined at the B3LYP level with different basis sets. Results show that the BEs of these GnH+⋯Mx+/2+ complexes will change into negative values when n ⩾ 4 for GnH+Lix+, n ⩾ 5 for GnH+Nax+, n ⩾ 6 for GnH+Kx+, n ⩾ 2 for GnH+Mgx2+, and n ⩾ 3 for GnH+Cax2+, respectively. The BEs of both G2H+Bex2+ and G3H+Bex2+, however, are always negative values. The signs “n” and “x” denote the number of glycine residues of linking the two cations (H+ and M+/2+) and the serial number of site bound by a metal-ion, respectively. Moreover, the BEs (ΔE) decrease gradually along the increase of oligoglycine size (n) between two cations for all these different GnH+Mx+/2+ systems, i.e., ΔE(GH+Mx+/2+) > ΔE(G2H+Mx+/2+) > ΔE(G3H+Mx+/2+). Monohydration at the metal-ion almost keeps these BEs unchangeable. The continuum solvent effect, however, can change them into negative values. Interestingly, the different potential well depths (or activation energy) on the potential energy surface give different information for these monovalent and divalent metal-ion involved systems. In detail, electrostatic effect of monovalent metal-ion on the proton transfer of amino-terminus decreases stepwise along with the number increase of the glycine residues between two cations. Moreover, the well will disappear in the process of proton transfer assisted by a water molecule. Differently, the (N–)H of neighboring the metal-bound site is the most potential proton (hydrogen) to transfer in these divalent metal-ion involved systems.
Co-reporter:Baotao Kang, Hongqi Ai, Jin Yong Lee
Carbon (May 2017) Volume 116() pp:
Publication Date(Web):May 2017
DOI:10.1016/j.carbon.2017.01.068
Density functional theory calculations were carried out to investigate how single-atom vacancies affect the electronic and magnetic properties of three types of graphyne model systems. Our simulations demonstrate that a single-atom vacancy can lead to an in-plane structural rearrangement, which plays an important role in tuning the electronic structures. A dispersionless spin-polarized band was observed around the fermi level, inducing strong magnetism in the three graphyne models. The single-atom vacancy can induce a 1.1–1.3 μB magnetic moment into αGy and βGy, while this value increases significantly to about 1.8μB for γGy. This single-atom vacancy technique is a promising method of manipulating the electronic and magnetic properties of graphyne.