Co-reporter:Guilan Fan, Ruifang Li, Zhenfeng Shang, Xiufang Xu
Computational and Theoretical Chemistry 2017 Volume 1118(Volume 1118) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.comptc.2017.08.025
•A strong hydrogen bond between hypervalent iodine compounds and HF is found.•The hydrogen bonds between Ph(R)IF and HF are electrostatic-covalent in nature.•The stronger hydrogen bonds in complexes under study have higher covalency.•The favorable substituents enhance the electrostatic interaction of hydrogen bonds.•Altering oxidation states of the related atoms can help design new hydrogen bonds.For the first time, the intermolecular hydrogen bonding interactions between polyvalent iodine compounds and hydrogen fluoride (HF) have been studied using M06-2X/SDD-6-311++G(d, p) method. The computational results show that, compared with iodine(I) compound IF, both the iodine(III) compound Ph(Et)IF and the iodine(V) compound Ph(Et)3IF have strong hydrogen bonding interactions with HF and the binding energies are about -21.00 kcal/mol. Geometry analysis, natural bond orbital (NBO) analysis, energy decomposition analysis, electrostatic potential (ESP) analysis on molecular vdW surface and topological analysis for the bond critical points (BCPs) suggest that these strong interactions are not only electrostatic but also highly covalent in nature. In addition, our computational studies indicate that owing to the lower electronegativity of Cl and Br atoms relative to that of F atom, the compounds Ph(Et)ICl and Ph(Et)IBr have much weaker hydrogen bonding interactions with HF than Ph(Et)IF. Moreover, the electronic effects of substituents at the iodine center of the hypervalent iodine (III) compounds on the strength of the hydrogen bonds have also been studied and the results show that the alkyl groups such as methyl, ethyl, isopropyl and tert-butyl groups can enhance the hydrogen bonding interaction obviously relative to other types of substituents. And the hydrogen bond in complex Ph(iPr)IF⋯HF is very strong with binding energy of −22.44 kcal/mol.Download high-res image (122KB)Download full-size image
Co-reporter:Jie Ning;Xiufang Xu
Catalysis Letters 2015 Volume 145( Issue 6) pp:1331-1343
Publication Date(Web):2015 June
DOI:10.1007/s10562-015-1524-5
The ruthenium-Cp* picolyl-NHC complexes catalyzed transfer hydrogenation reaction has been investigated with the aid of density functional theory calculations at the B3LYP level of theory. Two kinds of “hydridic route” (inner sphere mechanism) and one “direct hydrogen transfer route” (Meerwein–Ponndorf–Verley mechanism) have been examined. From the results we conclude that the stepwise inner sphere mechanism would be favored from the energetic point of view. The hemilability of the picolyl group plays an important role in determining which mechanism the transfer hydrogenation reaction should be followed. And the effect of agostic interaction has also been discussed.
Co-reporter:Cun-Qin Lv, Jun Li, Kai-Cheng Ling, Zhen-Feng Shang, Gui-Chang Wang
Surface Science 2010 Volume 604(9–10) pp:779-787
Publication Date(Web):15 May 2010
DOI:10.1016/j.susc.2010.01.027
The adsorption and decomposition of methylamine on Ni(1 1 1), Ni(1 0 0), stepped Ni(1 1 1), and nitrogen atom modified Ni(1 0 0) (denoted N–Ni(1 0 0)) have been studied with the DFT–GGA method using the periodic slab models. The initial scissions of C–H, N–H and C–N bond are considered. The adsorption energies under the most stable configurations for the possible species and the activation energies for the possible initial elementary reactions involved are obtained in the present work. Through systematic exploring of the kinetics mechanism of methylamine decomposition on these four surfaces, it is found that the reactivity of these surfaces decreased with the order of stepped Ni(1 1 1) > Ni(1 0 0) > Ni(1 1 1) > N–Ni(1 0 0). This indicates that the reactivity is related to the openness of the surface, and the presence of nitrogen atom reduces the reactivity of the Ni(1 0 0). For the three reactions, the barriers decreased with the order of C–N > N–H > C–H on Ni(1 1 1) and Ni(1 0 0), whereas they decreased with the order of C–N > C–H > N–H on stepped Ni(1 1 1) and N–Ni(1 0 0).
Co-reporter:Bin Xing, Xian-Yong Pang, Gui-Chang Wang, Zhen-Feng Shang
Journal of Molecular Catalysis A: Chemical 2010 315(2) pp: 187-196
Publication Date(Web):
DOI:10.1016/j.molcata.2009.09.010
Co-reporter:Hongyan Ma, Wenge Xu, Zhenfeng Shang, Guichang Wang
Journal of Natural Gas Chemistry (January 2011) Volume 20(Issue 1) pp:34-40
Publication Date(Web):1 January 2011
DOI:10.1016/S1003-9953(10)60143-0
AbstractAdsorption of cyclohexene and its dehydrogenation intermediates on the nAu/Pt(100) (n = 0, 1, 2 means clean Pt, one monolayer and two layers of Au covered Pt surfaces, respectively.) has been investigated by self-consistent (GGA-PW91) density functional theory combined with periodic slab model. It is found that on the clean platinum, there are two kinds of favorable adsorption sites, i.e., hollow sites and bridge sites, and the adsorption energy at the hollow site is larger than that at the bridge site. However, on the Au/Pt and 2Au/Pt surfaces, there are three kinds of adsorption sites, and the adsorption energies are alike at both the bridge site and the top site. The magnitude order of the adsorption energies is as follows: clean Pt > Au/Pt > 2Au/Pt. The configurations of cyclohexene molecule have been distorted a little during the geometry optimizations. The lengths of C-M (M = Pt or Au, on the top layer of the slab) bonds are closely related to the corresponding adsorption energies.

![Naphtho[1,8-bc]pyran-9-carbonitrile, 2,3,7,8-tetraphenyl-](/data/chemimg/141000/1401051-00-0.png)
![Naphtho[1,8-bc]pyran-9-carbonitrile, 2,3,7,8-tetraphenyl-](/data/chemimg/141000/1401051-00-0_b.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)
























![Diphenanthro[2,1-m:2',1'-m']chryseno[1,2-c:7,8-c']dipicene](/data/chemimg/141400/1309375-11-8.png)
![Diphenanthro[2,1-m:2',1'-m']chryseno[1,2-c:7,8-c']dipicene](/data/chemimg/141400/1309375-11-8_b.png)




























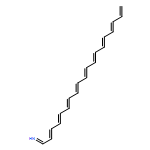

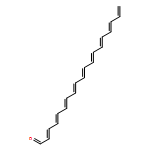
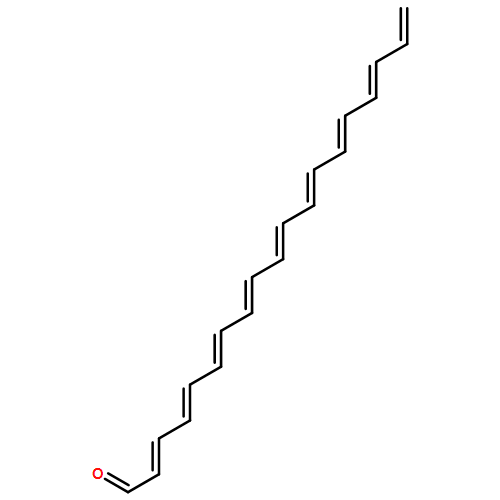
![Benzo[9,10]piceno[4,3-c]piceno[3,4-m]picene](/data/chemimg/105500/244064-73-1.png)
![Benzo[9,10]piceno[4,3-c]piceno[3,4-m]picene](/data/chemimg/105500/244064-73-1_b.png)


![Chryseno[1,2-c:7,8-c']dipicene](/data/chemimg/105500/154268-92-5.png)
![Chryseno[1,2-c:7,8-c']dipicene](/data/chemimg/105500/154268-92-5_b.png)
![Naphtho[1,2-c:5,6-c']dipicene](/data/chemimg/105500/154268-90-3.png)
![Naphtho[1,2-c:5,6-c']dipicene](/data/chemimg/105500/154268-90-3_b.png)
![Benzo[c]chryseno[2,1-m]picene](/data/chemimg/105500/147188-56-5.png)
![Benzo[c]chryseno[2,1-m]picene](/data/chemimg/105500/147188-56-5_b.png)
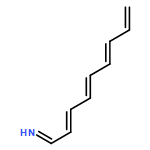


![Benzo[c]naphtho[2,1-m]picene](/data/chemimg/3743300/13547-46-1.png)
![Benzo[c]naphtho[2,1-m]picene](/data/chemimg/3743300/13547-46-1_b.png)





![Benzo[c]picene](http://img.cochemist.com/ccimg/300/217-37-8.png)
![Benzo[c]picene](http://img.cochemist.com/ccimg/300/217-37-8_b.png)