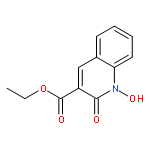
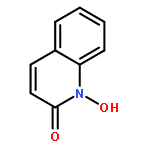
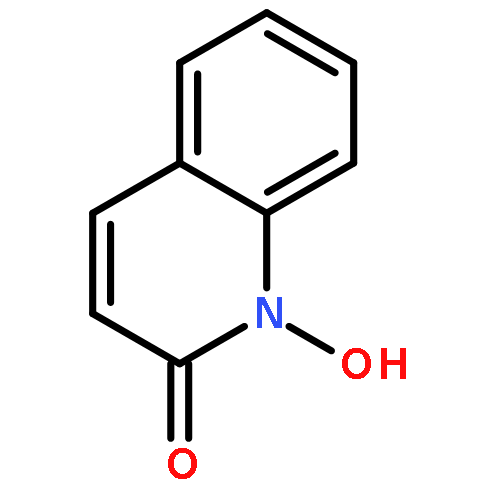
Co-reporter:Marvin J. Meyers, Elizabeth J. Anderson, Sarah A. McNitt, Thomas M. Krenning, Megh Singh, Jing Xu, Wentian Zeng, Limei Qin, Wanwan Xu, Siting Zhao, Li Qin, Christopher S. Eickhoff, Jonathan Oliva, Mary A. Campbell, Stacy D. Arnett, Michael J. Prinsen, David W. Griggs, Peter G. Ruminski, Daniel E. Goldberg, Ke Ding, Xiaorong Liu, et al.
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 16) pp:5144-5150
Publication Date(Web):15 August 2015
DOI:10.1016/j.bmc.2015.02.050
Given the rise of parasite resistance to all currently used antimalarial drugs, the identification of novel chemotypes with unique mechanisms of action is of paramount importance. Since Plasmodium expresses a number of aspartic proteases necessary for its survival, we have mined antimalarial datasets for drug-like aspartic protease inhibitors. This effort led to the identification of spiropiperidine hydantoins, bearing similarity to known inhibitors of the human aspartic protease β-secretase (BACE), as new leads for antimalarial drug discovery. Spiropiperidine hydantoins have a dynamic structure–activity relationship profile with positions identified as being tolerant of a variety of substitution patterns as well as a key piperidine N-benzyl phenol pharmacophore. Lead compounds 4e (CWHM-123) and 12k (CWHM-505) are potent antimalarials with IC50 values against Plasmodium falciparum 3D7 of 0.310 μM and 0.099 μM, respectively, and the former features equivalent potency on the chloroquine-resistant Dd2 strain. Remarkably, these compounds do not inhibit human aspartic proteases BACE, cathepsins D and E, or Plasmodium plasmepsins II and IV despite their similarity to known BACE inhibitors. Although the current leads suffer from poor metabolic stability, they do fit into a drug-like chemical property space and provide a new class of potent antimalarial agents for further study.
Co-reporter:Marvin J. Meyers, Micky D. Tortorella, Jing Xu, Limei Qin, Zhengxiang He, Xingfen Lang, Wentian Zeng, Wanwan Xu, Li Qin, Michael J. Prinsen, Francis M. Sverdrup, Christopher S. Eickhoff, David W. Griggs, Jonathan Oliva, Peter G. Ruminski, E. Jon Jacobsen, Mary A. Campbell, David C. Wood, Daniel E. Goldberg, Xiaorong Liu, Yongzhi Lu, Xin Lu, Zhengchao Tu, Xiaoyun Lu, Ke Ding, and Xiaoping Chen
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 1) pp:89-93
Publication Date(Web):December 6, 2013
DOI:10.1021/ml400412x
Given the threat of drug resistance, there is an acute need for new classes of antimalarial agents that act via a unique mechanism of action relative to currently used drugs. We have identified a set of druglike compounds within the Tres Cantos Anti-Malarial Set (TCAMS) which likely act via inhibition of a Plasmodium aspartic protease. Structure–activity relationship analysis and optimization of these aminohydantoins demonstrate that these compounds are potent nanomolar inhibitors of the Plasmodium aspartic proteases PM-II and PM-IV and likely one or more other Plasmodium aspartic proteases. Incorporation of a bulky group, such as a cyclohexyl group, on the aminohydantion N-3 position gives enhanced antimalarial potency while reducing inhibition of human aspartic proteases such as BACE. We have identified compound 8p (CWHM-117) as a promising lead for optimization as an antimalarial drug with a low molecular weight, modest lipophilicity, oral bioavailability, and in vivo antimalarial activity in mice.Keywords: aminohydantoin; antimalarial; aspartic protease inhibitors; Malaria; medicinal chemistry;
Co-reporter:Marvin J. Meyers, Scott A. Long, Matthew J. Pelc, Jane L. Wang, Scott J. Bowen, Mark C. Walker, Barbara A. Schweitzer, Heather M. Madsen, Ruth E. Tenbrink, Joseph McDonald, Sarah E. Smith, Susan Foltin, David Beidler, Atli Thorarensen
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6538-6544
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.08.055
Herein we report the identification of two new fatty acid amide hydrolase (FAAH) inhibitor lead series with FAAH kinact/Ki potency values greater than 1500 M−1 s−1. The two novel spirocyclic cores, 7-azaspiro[3.5]nonane and 1-oxa-8-azaspiro[4.5]decane, clearly distinguished themselves from the other spirocyclic cores on the basis of their superior potency for FAAH. Lead compounds from these two series have suitable FAAH potency and selectivity for additional medicinal chemistry optimization.
Co-reporter:Marvin J. Meyers, Scott A. Long, Matthew J. Pelc, Jane L. Wang, Scott J. Bowen, Barbara A. Schweitzer, Mark V. Wilcox, Joseph McDonald, Sarah E. Smith, Susan Foltin, Jeanne Rumsey, Young-Sun Yang, Mark C. Walker, Satwik Kamtekar, David Beidler, Atli Thorarensen
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6545-6553
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.08.048
Fatty acid amide hydrolase (FAAH) is an integral membrane serine hydrolase responsible for the degradation of fatty acid amide signaling molecules such as endocannabinoid anandamide (AEA), which has been shown to possess cannabinoid-like analgesic properties. Herein we report the optimization of spirocyclic 7-azaspiro[3.5]nonane and 1-oxa-8-azaspiro[4.5]decane urea covalent inhibitors of FAAH. Using an iterative design and optimization strategy, lead compounds were identified with a remarkable reduction in molecular weight and favorable CNS drug like properties. 3,4-Dimethylisoxazole and 1-methyltetrazole were identified as superior urea moieties for this inhibitor class. A dual purpose in vivo efficacy and pharmacokinetic screen was designed to be the key decision enabling experiment affording the ability to move quickly from compound synthesis to selection of preclinical candidates. On the basis of the remarkable potency, selectivity, pharmacokinetic properties and in vivo efficacy, PF-04862853 (15p) was advanced as a clinical candidate.
Co-reporter:Marvin J. Meyers ; Graciela B. Arhancet ; Susan L. Hockerman ; Xiangyang Chen ; Scott A. Long ; Matthew W. Mahoney ; Joseph R. Rico ; Danny J. Garland ; James. R. Blinn ; Joe T. Collins ; Shengtian Yang ; Horng-Chih Huang ; Kevin F. McGee ; Jay M. Wendling ; Jessica D. Dietz ; Maria A. Payne ; Bruce L. Homer ; Marcia I. Heron ; David B. Reitz ;Xiao Hu
Journal of Medicinal Chemistry 2010 Volume 53(Issue 16) pp:5979-6002
Publication Date(Web):July 30, 2010
DOI:10.1021/jm100505n
We have discovered a novel class of nonsteroidal pyrazoline antagonists of the mineralocorticoid receptor (MR) that show excellent potency and selectivity against other nuclear receptors. Early analogues were poorly soluble and had a propensity to inhibit the hERG channel. Remarkably, both of these challenges were overcome by incorporation of a single carboxylate moiety. Structural modification of carboxylate-containing lead R-4g with a wide range of substituents at each position of the pyrazoline ring resulted in R-12o, which shows excellent activity against MR and reasonable pharmacokinetic profile. Introduction of conformational restriction led to a novel series characterized by exquisite potency and favorable steroid receptor selectivity and pharmacokinetic profile. Oral dosing of 3S,3aR-27d (PF-3882845) in the Dahl salt sensitive preclinical model of salt-induced hypertension and nephropathy showed blood pressure attenuation significantly greater than that with eplerenone, reduction in urinary albumin, and renal protection. As a result of these findings, 3S,3aR-27d was advanced to clinical studies.
Co-reporter:Catherine W. Cai, Elena Lomonosova, Eileen A. Moran, Xiaohong Cheng, Kunjan B. Patel, Fabrice Bailly, Philippe Cotelle, Marvin J. Meyers, John E. Tavis
Antiviral Research (August 2014) Volume 108() pp:48-55
Publication Date(Web):August 2014
DOI:10.1016/j.antiviral.2014.05.007

![7H-Pyrido[2,1-b][1,3]thiazepine-7-carboxylicacid, octahydro-4-[[(2S)-2-mercapto-1-oxo-3-phenylpropyl]amino]-5-oxo-,(4S,7S,10aS)-](http://img.cochemist.com/ccimg/167400/167305-00-2.png)
![7H-Pyrido[2,1-b][1,3]thiazepine-7-carboxylicacid, octahydro-4-[[(2S)-2-mercapto-1-oxo-3-phenylpropyl]amino]-5-oxo-,(4S,7S,10aS)-](http://img.cochemist.com/ccimg/167400/167305-00-2_b.png)
![1H-Azepine-1-aceticacid,hexahydro-6-[[(2S)-2-mercapto-1-oxo-3-phenylpropyl]amino]-2,2-dimethyl-7-oxo-,(6S)-](http://img.cochemist.com/ccimg/160200/160135-92-2.png)
![1H-Azepine-1-aceticacid,hexahydro-6-[[(2S)-2-mercapto-1-oxo-3-phenylpropyl]amino]-2,2-dimethyl-7-oxo-,(6S)-](http://img.cochemist.com/ccimg/160200/160135-92-2_b.png)
![(4S,7S,12bR)-7-[2(S)-(Acetylsulfanyl)-3-phenylpropionamido]-6-oxo-1,2,3,4,6,7,8,12b-octahydropyrido[2,1-a][2]benzazepine-4-carboxylic acid](http://img.cochemist.com/ccimg/142700/142695-08-7.png)
![(4S,7S,12bR)-7-[2(S)-(Acetylsulfanyl)-3-phenylpropionamido]-6-oxo-1,2,3,4,6,7,8,12b-octahydropyrido[2,1-a][2]benzazepine-4-carboxylic acid](http://img.cochemist.com/ccimg/142700/142695-08-7_b.png)
![Cyclohexanecarboxylicacid,4-[[[1-[(2S)-3-[(2,3-dihydro-1H-inden-5-yl)oxy]-2-[(2-methoxyethoxy)methyl]-3-oxopropyl]cyclopentyl]carbonyl]amino]-,cis-](http://img.cochemist.com/ccimg/123200/123122-55-4.png)
![Cyclohexanecarboxylicacid,4-[[[1-[(2S)-3-[(2,3-dihydro-1H-inden-5-yl)oxy]-2-[(2-methoxyethoxy)methyl]-3-oxopropyl]cyclopentyl]carbonyl]amino]-,cis-](http://img.cochemist.com/ccimg/123200/123122-55-4_b.png)
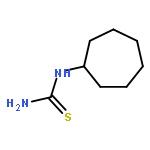























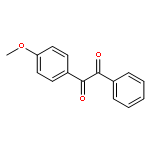
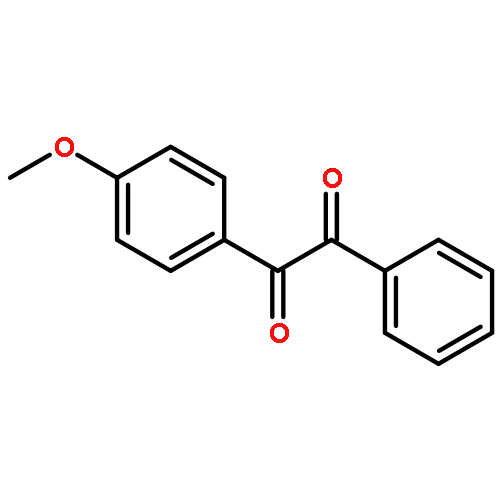
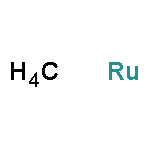







![Benzamide, N-[(cyclohexylamino)thioxomethyl]-](http://img.cochemist.com/ccimg/5000/4921-92-0.png)
![Benzamide, N-[(cyclohexylamino)thioxomethyl]-](http://img.cochemist.com/ccimg/5000/4921-92-0_b.png)