Co-reporter:Xiaocan Zhao and Liming Wang
The Journal of Physical Chemistry A May 4, 2017 Volume 121(Issue 17) pp:3247-3247
Publication Date(Web):April 10, 2017
DOI:10.1021/acs.jpca.7b00506
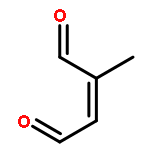
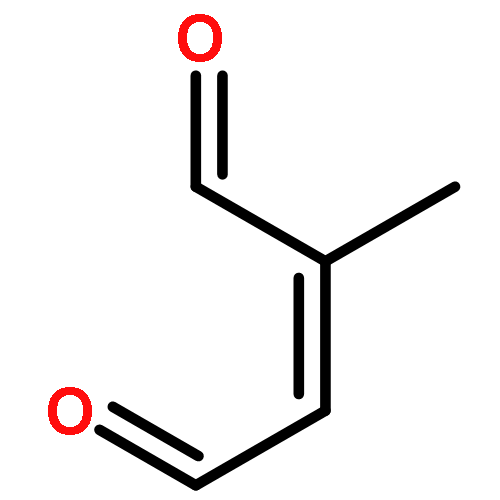
Furfural is emitted into the atmosphere because of its potential applications as an intermediate to alkane fuels from biomass, industrial usages, and biomass burning. The kinetic and mechanistic information on the furfural chemistry is necessary to assess the fate of furfural in the atmosphere and its impact on the air quality. Here we studied the atmospheric oxidation mechanisms of furfural initiated by the OH radicals using quantum chemistry and kinetic calculations. The reaction of OH and furfural was initiated mainly by OH additions to C2 and C5 positions, forming R2 and R5 adducts, which could undergo rapid ring-breakage to form R2B and R5B, respectively. Our calculations showed that these intermediate radicals reacted rather slowly with O2 under the atmospheric conditions because the additions of O2 to these radicals are only slightly exothermic and highly reversible. Alternatively, these radicals would react directly with O3, NO2, HO2/RO2, etc. Namely, the atmospheric oxidation of furfural would unlikely result in ozone formation. Under typical atmospheric conditions, the main products in OH-initiated furfural oxidation include 2-oxo-3-pentene-1,5-dialdehyde, 5-hydroxy-2(5H)-furanone, 4-oxo-2- butenoic acid, and 2,5-furandione. These compounds will likely stay in the gas phase and are subject to further photo-oxidation.
Co-reporter:Yi Yuan, Xiaocan Zhao, Sainan Wang, and Liming Wang
The Journal of Physical Chemistry A December 7, 2017 Volume 121(Issue 48) pp:9306-9306
Publication Date(Web):November 17, 2017
DOI:10.1021/acs.jpca.7b09741
The atmospheric oxidation mechanism of furan and methylfurans (MFs) initiated by OH radicals is studied using high-level quantum chemistry and kinetic calculations. The reaction starts mainly with OH addition to the C2/C5-position, forming highly chemically activated adduct radical R2*/R5*, which would either be stabilized by collision or promptly isomerize to R2B*/R5B* by breaking the C2-O/C5-O bond and then isomerize to other conformers of R2B/R5B by internal rotations. Under the atmospheric conditions, the ring-retaining radical R2/R5 would recombine with O2 and be converted to a 5-hydroxy-2-furanone compound and a compound containing epoxide, ester, and carbonyl functional groups, while the ring-opening radicals R2B/R5B would react with O2 and form unsaturated 1,4-dicarbonyl compounds. RRKM-ME calculations on the fate of R2*/R5* from the addition of OH and furans predict that the fractions of R2B/R5B formation, i.e., the molar yields of the corresponding dicarbonyl compounds, are 0.73, 0.43, 0.26, 0.07, and 0.28 for furan, 2-MF, 3-MF, 2,3-DMF, and 2,5-DMF, respectively, at 298 K and 760 Torr when using the RHF-UCCSD(T)-F12a/cc-pVDZ-F12 reaction energies and barrier heights. The predicted yields for dicarbonyl compounds agree reasonably with recent experimental measurements. Calculations here also suggest high yields of ring-retaining 5-hydroxy-2-furanone compounds, which might deserve further study.
Co-reporter:Sainan Wang and Liming Wang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 11) pp:7707-7714
Publication Date(Web):11 Feb 2016
DOI:10.1039/C5CP07199B
The atmospheric oxidation mechanisms of dimethyl ether (DME), diethyl ether (DEE) and diisopropyl ether (DiPE) are studied by using quantum chemistry and unimolecular reaction theory (RRKM-ME) calculations. For the peroxy radical CH3OCH2O2˙ from DME, a barrier height of ∼85 kJ mol−1 is found for its intramolecular H-shift to ˙CH2OCH2OOH, which can recombine rapidly with the atmospheric O2. RRKM-ME calculations obtain an effective rate of ∼0.1 s−1 at 298 K for the formation of ˙O2CH2OCH2OOH. For similar radicals in DEE and DiPE, effective rates are 1.6 s−1 and 1.1 s−1, respectively. In the atmosphere, these unimolecular reactions are fast enough to compete with the bimolecular reactions with NO and/or HO2, especially when [NO] is low. The fates of radicals after the H-shifts are also examined here. Several subsequent reactions are found to recycle OH radicals. New mechanisms are proposed on the basis of present calculations and are consistent with previous experimental results. In the atmosphere, the routes via H-shifts represent an auto-oxidation of these ethers with no involvement of NOx and therefore no O3 formation, and also a self-cleaning mechanism of organic compounds due to recycling of OH radicals. Some of the end products are highly oxidized with multifunctional groups and high O:C ratios, suggesting their low volatility and potential contribution to secondary organic aerosols.
Co-reporter:Runrun Wu, Yun Li, Shanshan Pan, Sainan Wang and Liming Wang
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 36) pp:23413-23422
Publication Date(Web):12 Aug 2015
DOI:10.1039/C5CP02731D
The atmospheric oxidation mechanism of 2-methylnaphthalene (2-MN) initiated by OH radicals is investigated by using quantum chemistry at BH&HLYP/6-311++G(2df,2p) and ROCBS-QB3 levels and kinetic calculations by transient state theory and unimolecular reaction theory coupled with master equation (RRKM-ME). This reaction is mainly initiated by OH additions, forming adducts Rn (2-MN-n-OH, n = 1–8). The fates of R1 and R3, representing the α- and β-adducts, are examined. The fates of R1 and R3 are found to be drastically different. In the atmosphere, R1 reacts with O2via O2 addition to the C2 position to form R1-2OO-a/s, which will undergo a bimolecular reaction with the atmospheric NO or unimolecular isomerization via intramolecular H-shifts, of which the latter is found to be dominant and accounts for the formation of dicarbonyl compounds observed in experimental studies. The role of the tricyclic radical intermediates formed from the ring-closure of R1-2OO is rather limited because their formation is endothermic and reversible, being contrary to the important role of the analogous bicyclic radical intermediates in the oxidation of benzenes. On the other hand, the fate of R3 is similar to that of the benzene–OH adduct, and the tricyclic intermediates will play an important role. An oxidation mechanism is proposed based on the theoretical predictions, and the routes for the experimentally observed products are suggested and compared.
Co-reporter: Liming Wang
ChemPhysChem 2015 Volume 16( Issue 7) pp:1542-1550
Publication Date(Web):
DOI:10.1002/cphc.201500012
Abstract
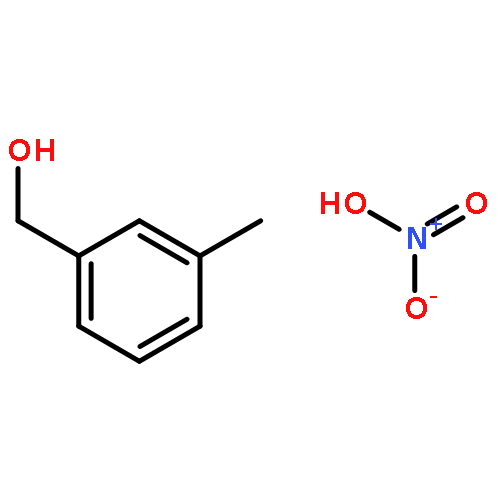
Benzyl alcohol (BA) is present in indoor atmospheres, where it reacts with OH radicals and undergoes further oxidation. A theoretical study is carried out to elucidate the reaction mechanism and to identify the main products of the oxidation of BA that is initiated by OH radicals. The reaction is found to proceed by H-abstraction from the CH2 group (25 %) and addition to the ipso (60 %) and ortho (15 %) positions of the aromatic ring. The BA–OH adducts react further with O2 via the bicyclic radical intermediates—the same way as for benzene—forming mainly 3-hydroxy-2-oxopropanal and butenedial. If NOx is low, the bicyclic peroxy radicals undergo intramolecular H-migration, forming products containing OH, OOH, and CH2OH/CHO functional groups, and contribute to secondary organic aerosol (SOA) formation.
Co-reporter:Runrun Wu, Sainan Wang, and Liming Wang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 1) pp:112-117
Publication Date(Web):December 8, 2014
DOI:10.1021/jp511616j
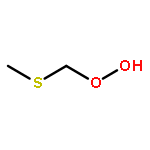
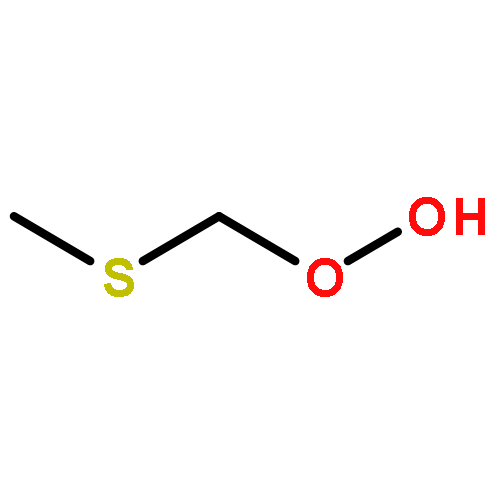
Theoretical study has been carried out on the fate of methylthiomethylperoxy radical (CH3SCH2OO, MSP) in the atmosphere. The intramolecular H-shift followed by recombination with O2, MSP → CH2SCH2OOH → OOCH2SCH2OOH (MSPO2), is found to be fast enough, that is, 2.1 s–1 at 293 K, to compete with and even surpass the possible bimolecular reactions of MSP with NOx, HO2, and RO2 in the remote marine atmosphere. MSPO2 would also undergo another intramolecular H-shift and decompose to the most important intermediate HOOCH2SCHO instead of the CH3SCH2O radical. HOOCH2SCHO would be further oxidized via the route as HOOCH2SCO (by OH radical) → HOOCH2S (by decomposition) → HOOCH2SO (by O3 or NO2) → HOOCH2SO2 (by O3 and NO2) → OH + CH2O + SO2 (by decomposition). Our calculations suggest a drastically different oxidation mechanism for dimethyl sulfide (CH3SCH3, DMS) in the remote marine atmosphere.
Co-reporter:Yun Li and Liming Wang
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 33) pp:17908-17917
Publication Date(Web):14 Jul 2014
DOI:10.1039/C4CP02027H
The atmospheric oxidation mechanism of 1,2,4-trimethylbenzene (1,2,4-TMB) initiated by OH radicals is investigated using quantum chemistry calculations at M06-2X and ROCBS-QB3 levels. The calculations show that the initiation of the reaction is dominated by OH addition to C1, C3 and C5 to form 1,2,4-TMB-OH adducts R1, R3, and R5 with branching ratios of 0.22, 0.19, and 0.38, respectively, using ROCBS-QB3 energies. In the troposphere, the adducts react with O2 by irreversible H-abstraction to form phenolic compounds and by reversible addition to TMB-OH-O2 peroxy radicals, which will cyclize to bicyclic radicals, similar to those in benzene, toluene, and xylenes. The bicyclic radicals can further recombine with O2 to generate bicyclic peroxy and alkoxyl radicals. The bicyclic alkoxyl radicals would break the ring directly to form 1,2-dicarbonyl products and unsaturated 1,4-dicarbonyl co-products, or undergo another cyclization to form an epoxy group, followed by the ring-breakage to form 1,2-dicarbonyl products and epoxy-1,4-dicarbonyl co-products. The predicted yields of products agree reasonably with the previous experimental measurements, while considerable discrepancies also exist for the yields of nitrates, biacetyl, 4-oxo-2-pentenal, and butenedial, etc. Our mechanism also predicts a new type of epoxy-1,4-dicarbonyl compounds with a total yield of ∼0.32. The epoxy-1,4-dicarbonyl compounds have not been suggested or reported in previous studies.
Co-reporter:Shanshan Pan and Liming Wang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10778-10787
Publication Date(Web):October 16, 2014
DOI:10.1021/jp506815v
The atmospheric oxidation mechanism of m-xylene (mX) initiated by the OH radical is investigated at M06-2X and ROCBS-QB3 levels, coupled with reaction kinetics calculations by using transition state theory and unimolecular RRKM-ME theory. The calculations show that the reaction between OH and mX is dominated by OH addition to the C2 and C4 positions, forming adducts mX–2-OH (R2) and mX–4-OH (R4). In the atmosphere, R2 and R4 react with O2 by irreversible H-abstraction to dimethylphenols or by reversible additions to bicyclic radical intermediates, which would recombine again with O2 to form bicyclic peroxy radicals, to bicyclic alkoxyl radicals by reacting with NO or HO2, and eventually to final products such as glyoxal, methylglyoxal, and their coproducts. The effects of reaction pressure and temperature are explored by RRKM-ME calculations. A mechanism at 298 K is proposed on the basis of current predictions and previous experimental and modeling results. The predicted product yields support the values in the SAPRC mechanism, even though the predicted yield of 1.0% for glyoxal is lower than the value of ∼11% from the experimental measurements.
Co-reporter:Runrun Wu, Shanshan Pan, Yun Li, and Liming Wang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 25) pp:4533-4547
Publication Date(Web):June 5, 2014
DOI:10.1021/jp500077f
The atmospheric oxidation mechanism of toluene initiated by OH radical addition is investigated by quantum chemistry calculations at M06-2X, G3MP2-RAD, and ROCBS-QB3 levels and by kinetics calculation by using transition state theory and unimolecular reaction theory coupled with master equation (RRKM-ME). The predicted branching ratios are 0.15, 0.59, 0.05, and 0.14 for OH additions to ipso, ortho, meta, and para positions (forming R1–R4 adducts), respectively. The fate of R2, R4, and R1 is investigated in detail. In the atmosphere, R2 reacts with O2 either by irreversible H-abstraction to form o-cresol (36%), or by reversible recombination to R2-1OO-syn and R2-3OO-syn, which subsequently cyclize to bicyclic radical R2-13OO-syn (64%). Similarly, R4 reacts with O2 with branching ratios of 61% for p-cresol and 39% for R4-35OO-syn, while reaction of R1 and O2 leads to R1-26OO-syn. RRKM-ME calculations show that the reactions of R2/R4 with O2 have reached their high-pressure limits at 760 Torr and the formation of R2-16O-3O-s is only important at low pressure, i.e., 5.4% at 100 Torr. The bicyclic radicals (R2-13OO-syn, R4-35OO-syn, and R1-26OO-syn) will recombine with O2 to produce bicyclic alkoxy radicals after reacting with NO. The bicyclic alkoxy radicals would break the ring to form products methylglyoxal/glyoxal (MGLY/GLY) and their corresponding coproducts butenedial/methyl-substituted butenedial as proposed in earlier studies. However, a new reaction pathway is found for the bicyclic alkoxy radicals, leading to products MGLY/GLY and 2,3-epoxybutandial/2-methyl-2,3-epoxybutandial. A new mechanism is proposed for the atmospheric oxidation mechanism of toluene based on current theoretical and previous theoretical and experimental results. The new mechanism predicts much lower yield of GLY and much higher yield of butenedial than other atmospheric models and recent experimental measurements. The new mechanism calls for detection of proposed products 2,3-epoxybutandial and 2-methyl-2,3-epoxybutandial.
Co-reporter:Liming Wang, Runrun Wu, and Cui Xu
The Journal of Physical Chemistry A 2013 Volume 117(Issue 51) pp:14163-14168
Publication Date(Web):December 2, 2013
DOI:10.1021/jp4101762
The fate of alkoxy radicals formed in the atmospheric oxidation of benzene initiated by OH radical is investigated by using quantum chemistry and kinetics calculations. The two alkoxy radicals (R2 and R3), formed from the commonly accepted bicyclic radical intermediates, are found to undergo ring-closure preferentially, in addition to the ring-breakage, as suggested in previous studies. The ratio between the ring-closure and ring-breakage is ∼2:1. The ring-closure route will lead to equal amounts of glyoxal and 2,3-epoxybutandial, while the ring-breakage route leads to glyoxal and butenedial. Overall, the new mechanism suggests the yield of glyoxal to be three times that of butenedial, consistent with the previous experimental measurements. The new mechanism calls for the search of the newly proposed product 2,3-epoxybutandial.
Co-reporter:Cui Xu and Liming Wang
The Journal of Physical Chemistry A 2013 Volume 117(Issue 11) pp:2358-2364
Publication Date(Web):February 26, 2013
DOI:10.1021/jp308856b
The gas-phase oxidation mechanism of phenol initiated by OH radical was investigated using DFT and ab initio calculations. The initiation of the reaction is dominated by OH addition to ortho-position, forming P2, which subsequently combines with O2 at the ipso-position to form P2-1-OO adduct. A concerted HO2 elimination process from P2-1-OO was found to be much faster than the common ring closure to bicyclic intermediates. The HO2 elimination process from P2-1-OO forms 2-hydroxy-3,5-cyclohexadienone (HCH) as the main product and is also responsible for the experimental fact that the rate constants for reaction between P2 and O2 are about 2 orders of magnitude higher than those between other aromatic–OH adducts and O2. It was speculated that HCH would isomerize to catechol, which is thermodynamically more stable than HCH and was the experimentally observed main product, possibly through heterogeneous processes. Reaction of P2 with NO2 proceeded by addition to form P2-n-NO2 (n = 1, 3, 5), followed by HONO elimination from P2-1/3-NO2 to form catechol. The barriers for HONO elimination and catechol formation are below the separate reactants P2 and NO2, being consistent with the experimental observation of catechol in the absence of O2, while H2O elimination from P2-1/3-NO2 to form 2-nitrophenol (2NP) is hindered by high barriers. The most likely pathway for 2NP is the reaction of phenoxy radical and NO2.
Co-reporter:Zhijie Zhang, Xiaoyan Xu, and Liming Wang
The Journal of Physical Chemistry A 2013 Volume 117(Issue 1) pp:160-168
Publication Date(Web):December 10, 2012
DOI:10.1021/jp309505s
The atmospheric oxidation mechanism of 2,7-dimethylnaphthalene (27DMN) initiated by OH radical is investigated at levels of BB1K and G3MP2-RAD//BH&HLYP. The reaction is mainly initiated by OH addition to the C1 position to form radical adduct R1. In the atmosphere, R1 reacts with O2 via two comparable pathways as direct H abstraction to form 27DMN-1-ol and as O2 addition to the C2 position to form R1-2OO radicals, both being slow with rate constants of 10–18–10–17 cm3 molecule–1 s–1. The R1-2OO-s conformer is found to be important in 27DMN oxidation whereas the role of the R1-2OO-a conformer is negligible. Radicals R1-2OO-s have three comparable pathways: ring closure to tricyclic intermediate R1-29OO-s, intramolecular H shift from −OH to −OO to form dicarbonyl products, and reactions with atmospheric NO and/or HO2, etc. The ring closure to R1-29OO-s is endothermic and reversible whereas similar ring closures in benzene and toluene oxidations are exothermic and irreversible. The intramolecular H shift becomes prominent because of the reversibility of ring closure in 27DMN oxidation and is responsible for rapid formation of dicarbonyl compound (C12H12O2) in simulation chamber studies. The oxy radical (R1-2O herein) would not undergo C1–C2 cleavage to form dicarbonyl, as suggested in previous studies; instead, R1-2O would close the ring to form epoxide radical R1-23O. Radical R1-29OO-s would recombine with the atmospheric O2 and isomerize to diepoxide radical R1-23O-89O-s at comparable rates, and study on their further reactions is desirable.
Co-reporter:Zhijie Zhang, Ling Lin and Liming Wang
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 8) pp:2645-2650
Publication Date(Web):07 Dec 2011
DOI:10.1039/C2CP23271E
The atmospheric oxidation mechanism of naphthalene (Nap) initiated by the OH radical is investigated using density functional theory at B3LYP and BB1K levels. The initial step is dominated by OH addition to the C1-position of Nap, forming radical C10H8-1-OH (R1), followed by the O2 additions to the C2 position to form peroxy radical R1-2OO, or by the hydrogen abstraction by O2 to form 1-naphthol. In the atmosphere, R1-2OO will react with NO to form R1-2O, undergo intramolecular hydrogen transfer from –OH to –OO to form R1-P2O1 radicals, or possibly undergo ring-closure to R1-29OO bi-cyclic radical; while the formation of other bi-cyclic intermediate radicals is negligible because of the extremely high Gibbs energy barriers of >100 kJ mol−1 (relative to R1+O2). The mechanism is different from the oxidation mechanism of benzene, where the bi-cyclic intermediates play an important role. Radicals R1-P2O1 will dissociate to 2-formylcinnamaldehyde, while R1-2O will be transformed to stable products C10H6O3via epoxide-like intermediates. A few reaction pathways suggested in previous experimental studies are found to be invalid.
Co-reporter:Liming Wang, Jingsong Zhang
International Journal of Mass Spectrometry 2012 Volume 311() pp:56-63
Publication Date(Web):1 February 2012
DOI:10.1016/j.ijms.2011.12.005
The structural and energetic information of SixGeyHz and ions is crucial in understanding the deposition processes in producing SixGe1−x semiconductor materials. This work presents theoretical studies on the structures and energetics of the simplest SiGe-hydrides and cations, SiGeHz0,+1, as well as Ge2Hz0,+1 and Si2Hz0,+1 for comparison. The structures are obtained at DFT-B3LYP and MP2 levels with 6-31+G(2df,p) basis set, and the electronic energies at Gaussian-4 (G4) level. The G4 energies are used to calculate the relative energies, bond dissociation energies, the adiabatic ionization energies (IEas) of neutral species, and the appearance energies (AEs) of cation fragments from SiGeH6, Ge2H6, and Si2H6. The relative energies and IEas for Si2Hz and the total atomization energies of Si2Hz and Ge2Hz are compared and are in close agreement with previous theoretical and experimental studies, while the agreements on the AEs of Si2Hz+ from Si2H6 are less pronounced. The calculations suggest that the kinetic shift effect and potential barriers should be taken into account when using AEs for thermodynamic information of Si2H2+, Ge2H2+ and SiGeH2+.Graphical abstractHighlights► Structures of SiGeHz and cations are examined by quantum chemistry calculations. ► Adiabatic ionization energies are predicted at G4 level. ► Appearance energies of ion fragments from SiGeH6 are predicted. ► Results are compared with those for Si2Hz and Ge2Hz. ► Photoionization of SiGeH6 is discussed with the potential energy surface of SiGeH6+.
Co-reporter:Liming Wang, Aili Tang
Chemical Physics 2011 Volume 382(1–3) pp:98-103
Publication Date(Web):28 April 2011
DOI:10.1016/j.chemphys.2011.03.006
Abstract
The reaction mechanisms for dimethyl selenide (DMSe) and dimethyl selenoxide (DMSeO) with OH radical are studied by using quantum chemistry calculations. The structures are optimized at MP2 levels, and electronic energies at Gaussian-4 level. Addition complexes are found between reactants as (CH3)2Se · OH, (CH3)2Se(O) · OH, and (CH3)2SeO · HO, which serve as reaction intermediates for further decompositions to CH3SeOH + CH3/CH3SeCH2 + H2O, CH3Se(O)OH + CH3, and CH3Se(O)CH2 + H2O, respectively. Under the atmospheric conditions, the dominant product channel for DMSe + OH is CH3SeCH2 and (CH3)2Se · OH, of which the latter is subsequently converted to DMSeO; while the main product for DMSeO + OH is CH3Se(O)OH. Hydrate formation for DMSeO would not alter the branch ratios because of its small fraction in the atmosphere.
Co-reporter:Aili Tang, Liming Wang, Ruiheng Zhou
Journal of Molecular Structure: THEOCHEM 2010 Volume 960(1–3) pp:31-39
Publication Date(Web):30 November 2010
DOI:10.1016/j.theochem.2010.08.021
Chlorinated benzoates are widely spread in the environment and are subject to anaerobic degradations, where chlorinated benzoates are used as electron acceptor by certain groups of anaerobic bacteria and the energy of the reduction reactions is used by bacteria for growth. The energies, namely the change of Gibbs energy of the reduction reaction (ΔrG), are predicted here using quantum chemistry calculations, where the gas-phase properties, including the enthalpies of formation, the Gibbs free energies, and the acidities, are predicted at the G3XMP2 level, and the solvent effects are modelled using the polarizable conductor model (C-PCM) model. The predicted gas-phase enthalpies of formation and acidities of monochlorinated benzoic acids are in excellent agreement with the experimental measurements, and the aqueous phase pKa prediction shows that chlorinated benzoic acids will exist as benzoates almost exclusively in the natural environment (pH ∼7).
Co-reporter:Liming Wang, Yanfen Liu
Journal of Molecular Structure: THEOCHEM 2010 Volume 957(1–3) pp:72-76
Publication Date(Web):15 October 2010
DOI:10.1016/j.theochem.2010.07.009
The enthalpies of formation of brominated benzenes and phenols were predicted using Gaussian-4 (G4), G3X, and G3XMP2 model chemistries and a few popular density functional methods, coupled with homodesmic reactions and in which C6H6, C6H5Br, and C6H5OH are used as reference compounds. The results from G4, G3X, and G3XMP2 agree closely within 2 kJ/mol for all brominated benzenes and phenols; while the results from density functional methods are systematically higher than the G4 ones. The predicted enthalpies of formation for 2- and 4-bromophenols are in close agreement with the recent experimental measurements. Three reactions , and were also used to derive ΔfH298K°(g, C6H5Br) = 98.7 ± 1.0 kJ/mol by using CH4, CH3Br, CH2Br2, CH2CHBr, and C6H6 as reference compounds at G4 and G3X levels. The value is significantly lower than the only experimental value of 105.4 kJ/mol given by Cox and Pilcher in 1970, and a re-determination is called.
Co-reporter:Yi-Liang He
Structural Chemistry 2009 Volume 20( Issue 3) pp:461-479
Publication Date(Web):2009 June
DOI:10.1007/s11224-009-9444-x
The DFT-B3LYP and G3X model chemistry were used to predict the cation structures and energetics of fluorinated, chlorinated, and brominated methanes. Ion–complex structures between methylene cations and HX (X = F, Cl, Br) were found for all H-containing cations, and [CHF–FH]+, [CF2–FH]+, [CCl2–ClH]+, and [CCl2–FH]+ structures are more stable than their normal tetravalent structures. Several cations should also be better described as ion–complex structures between methyl cations and halogen atoms, e.g., [CF3–Br]+. Transition states connecting normal and ion–complex structures were also located, and potential energy diagrams were constructed for decomposition of methane cations and to predict the fragmentation pathways. The G3X energies were used to predict the adiabatic ionization energies (IEas) and ion fragment appearance energies (AEs) from methanes. Many of the experimental AEs correspond to the energies of transition states instead of the thermodynamic dissociation limits.
Co-reporter:Yi-Liang He, Liming Wang
Journal of Molecular Structure: THEOCHEM 2009 Volume 913(1–3) pp:240-246
Publication Date(Web):15 November 2009
DOI:10.1016/j.theochem.2009.08.007
The proton affinities (PAs) and potential energy surfaces (PESs) of hydrochlorofluoromethanes (HCFMs) have been predicted by using Gaussian-3X (G3X) method. The G3X PAs agree with previous G3 predictions, while the large discrepancies between theoretical and experimental PAs persisted for CH2F2, CHF3, and CF3Cl. Protonated HCFMs usually have multiple structures, and structures with protonations at F-atom, [Methyl-FH]+, are the most stable. Transition states connecting different cation structures have been identified as proton exchanges between C–H σ-bonds or between halogen atoms. While the high transition barriers hinder the isomerization between different cation structures, protonated HCFMs from proton transfer reactions of HCFMs with HCFM+/HCO+/HN2+ may have high enough energy to decompose to methyl+ + HF/HCl or isomerize to less stable structures. Under low collision energy condition, reactions of methyl+ and H2/HF/HCl form ion complexes only, except for CH3+ + HCl, where CH2Cl+ + H2 can be formed by crossing over the transition barrier from [CH3–ClH]+ to [CH2Cl–H2]+.
Co-reporter:Liming Wang and Yi-Liang He
The Journal of Physical Chemistry A 2009 Volume 113(Issue 1) pp:238-245
Publication Date(Web):December 15, 2008
DOI:10.1021/jp802091z
The standard gas-phase enthalpies of formation of polychlorinated dibenzofurans (PCDFs) have been predicted by using G3XMP2 model chemistry, density functional theory (DFT), and second-order Muller−Plesset (MP2) theory, coupled with isodesmic reactions. The results show a large difference between G3XMP2 and DFT methods with 6-31G(2df,p) and 6-311++G(3df,3pd) basis sets, while MP2/G3MP2Large calculations agree closely with G3XMP2. Two isodesmic reaction schemes are used for better prediction of formation enthalpies. The first (IR1) employs monochlorobenzene as a reference species and the second (IR2) employs polychlorinated benzenes as reference species. The relative stability of PCDFs is rationalized by positional interactions. While the Cl-substitution at position 1/9 leads to the most stable isomers, the simultaneous substitutions at positions 1 and 9 result in a strong repulsion between Cl atoms. Failure of DFT-B3LYP is due to the overestimation of o-ClCl repulsion. For 1,9-PCDFs, the torsion motions of the benzene rings have extremely low harmonic vibrational frequencies. Their contributions to entropy, heat capacity, and thermal corrections have been calculated by using the numerically evaluated energy levels. The PCDF isomer patterns are also discussed based on the calculated thermodynamic parameters.
Co-reporter:Liming Wang
The Journal of Physical Chemistry A 2008 Volume 112(Issue 22) pp:4951-4957
Publication Date(Web):May 13, 2008
DOI:10.1021/jp0774443
The enthalpies of formation of stable closed shell C1 and C2 brominated hydrocarbons have been predicted using Gaussian-3X model chemistry. The entropy, heat capacity, and thermal corrections are calculated from B3LYP/6-31G(2df,p) geometries and vibrational frequencies using rigid-rotor−harmonic-oscillator approximation, except for the quantities of the internal rotations in ethanes, which are calculated using the quantum-mechanical energy levels. Enthalpies of formation have been obtained from G3X atomization and isodesmic reactions. Good agreement is observed on the well-established experimental enthalpies of formation of CH3Br, CH2Br2, CH2ClBr, and C2H3Br from the high-resolution threshold photoelectron photoionization coincidence study.
Co-reporter:Sainan Wang and Liming Wang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 11) pp:NaN7714-7714
Publication Date(Web):2016/02/11
DOI:10.1039/C5CP07199B
The atmospheric oxidation mechanisms of dimethyl ether (DME), diethyl ether (DEE) and diisopropyl ether (DiPE) are studied by using quantum chemistry and unimolecular reaction theory (RRKM-ME) calculations. For the peroxy radical CH3OCH2O2˙ from DME, a barrier height of ∼85 kJ mol−1 is found for its intramolecular H-shift to ˙CH2OCH2OOH, which can recombine rapidly with the atmospheric O2. RRKM-ME calculations obtain an effective rate of ∼0.1 s−1 at 298 K for the formation of ˙O2CH2OCH2OOH. For similar radicals in DEE and DiPE, effective rates are 1.6 s−1 and 1.1 s−1, respectively. In the atmosphere, these unimolecular reactions are fast enough to compete with the bimolecular reactions with NO and/or HO2, especially when [NO] is low. The fates of radicals after the H-shifts are also examined here. Several subsequent reactions are found to recycle OH radicals. New mechanisms are proposed on the basis of present calculations and are consistent with previous experimental results. In the atmosphere, the routes via H-shifts represent an auto-oxidation of these ethers with no involvement of NOx and therefore no O3 formation, and also a self-cleaning mechanism of organic compounds due to recycling of OH radicals. Some of the end products are highly oxidized with multifunctional groups and high O:C ratios, suggesting their low volatility and potential contribution to secondary organic aerosols.
Co-reporter:Yun Li and Liming Wang
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 33) pp:NaN17917-17917
Publication Date(Web):2014/07/14
DOI:10.1039/C4CP02027H
The atmospheric oxidation mechanism of 1,2,4-trimethylbenzene (1,2,4-TMB) initiated by OH radicals is investigated using quantum chemistry calculations at M06-2X and ROCBS-QB3 levels. The calculations show that the initiation of the reaction is dominated by OH addition to C1, C3 and C5 to form 1,2,4-TMB-OH adducts R1, R3, and R5 with branching ratios of 0.22, 0.19, and 0.38, respectively, using ROCBS-QB3 energies. In the troposphere, the adducts react with O2 by irreversible H-abstraction to form phenolic compounds and by reversible addition to TMB-OH-O2 peroxy radicals, which will cyclize to bicyclic radicals, similar to those in benzene, toluene, and xylenes. The bicyclic radicals can further recombine with O2 to generate bicyclic peroxy and alkoxyl radicals. The bicyclic alkoxyl radicals would break the ring directly to form 1,2-dicarbonyl products and unsaturated 1,4-dicarbonyl co-products, or undergo another cyclization to form an epoxy group, followed by the ring-breakage to form 1,2-dicarbonyl products and epoxy-1,4-dicarbonyl co-products. The predicted yields of products agree reasonably with the previous experimental measurements, while considerable discrepancies also exist for the yields of nitrates, biacetyl, 4-oxo-2-pentenal, and butenedial, etc. Our mechanism also predicts a new type of epoxy-1,4-dicarbonyl compounds with a total yield of ∼0.32. The epoxy-1,4-dicarbonyl compounds have not been suggested or reported in previous studies.
Co-reporter:Zhijie Zhang, Ling Lin and Liming Wang
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 8) pp:NaN2650-2650
Publication Date(Web):2011/12/07
DOI:10.1039/C2CP23271E
The atmospheric oxidation mechanism of naphthalene (Nap) initiated by the OH radical is investigated using density functional theory at B3LYP and BB1K levels. The initial step is dominated by OH addition to the C1-position of Nap, forming radical C10H8-1-OH (R1), followed by the O2 additions to the C2 position to form peroxy radical R1-2OO, or by the hydrogen abstraction by O2 to form 1-naphthol. In the atmosphere, R1-2OO will react with NO to form R1-2O, undergo intramolecular hydrogen transfer from –OH to –OO to form R1-P2O1 radicals, or possibly undergo ring-closure to R1-29OO bi-cyclic radical; while the formation of other bi-cyclic intermediate radicals is negligible because of the extremely high Gibbs energy barriers of >100 kJ mol−1 (relative to R1+O2). The mechanism is different from the oxidation mechanism of benzene, where the bi-cyclic intermediates play an important role. Radicals R1-P2O1 will dissociate to 2-formylcinnamaldehyde, while R1-2O will be transformed to stable products C10H6O3via epoxide-like intermediates. A few reaction pathways suggested in previous experimental studies are found to be invalid.
Co-reporter:Runrun Wu, Yun Li, Shanshan Pan, Sainan Wang and Liming Wang
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 36) pp:NaN23422-23422
Publication Date(Web):2015/08/12
DOI:10.1039/C5CP02731D
The atmospheric oxidation mechanism of 2-methylnaphthalene (2-MN) initiated by OH radicals is investigated by using quantum chemistry at BH&HLYP/6-311++G(2df,2p) and ROCBS-QB3 levels and kinetic calculations by transient state theory and unimolecular reaction theory coupled with master equation (RRKM-ME). This reaction is mainly initiated by OH additions, forming adducts Rn (2-MN-n-OH, n = 1–8). The fates of R1 and R3, representing the α- and β-adducts, are examined. The fates of R1 and R3 are found to be drastically different. In the atmosphere, R1 reacts with O2via O2 addition to the C2 position to form R1-2OO-a/s, which will undergo a bimolecular reaction with the atmospheric NO or unimolecular isomerization via intramolecular H-shifts, of which the latter is found to be dominant and accounts for the formation of dicarbonyl compounds observed in experimental studies. The role of the tricyclic radical intermediates formed from the ring-closure of R1-2OO is rather limited because their formation is endothermic and reversible, being contrary to the important role of the analogous bicyclic radical intermediates in the oxidation of benzenes. On the other hand, the fate of R3 is similar to that of the benzene–OH adduct, and the tricyclic intermediates will play an important role. An oxidation mechanism is proposed based on the theoretical predictions, and the routes for the experimentally observed products are suggested and compared.

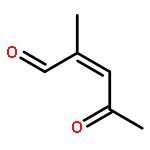



![Naphth[2,3-b]oxirene-2,7-dione,1a,7a-dihydro-1a-methyl-](http://img.cochemist.com/ccimg/15500/15448-59-6.png)
![Naphth[2,3-b]oxirene-2,7-dione,1a,7a-dihydro-1a-methyl-](http://img.cochemist.com/ccimg/15500/15448-59-6_b.png)