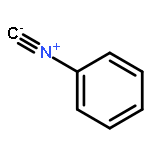
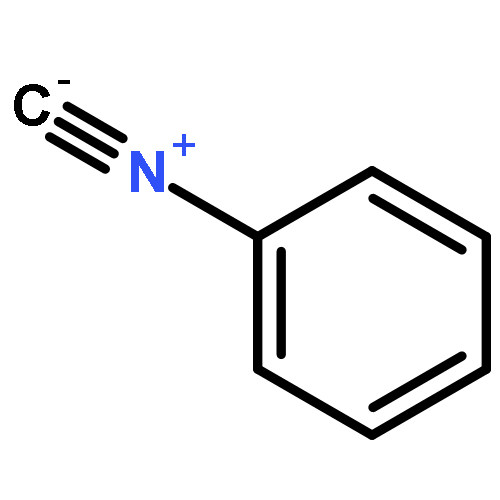
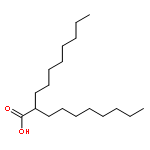
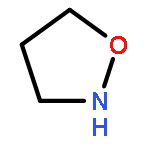
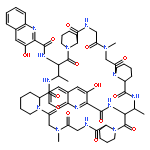
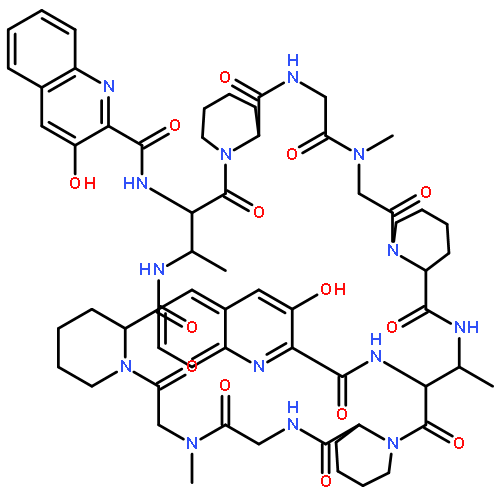
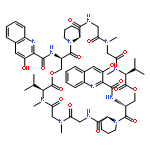
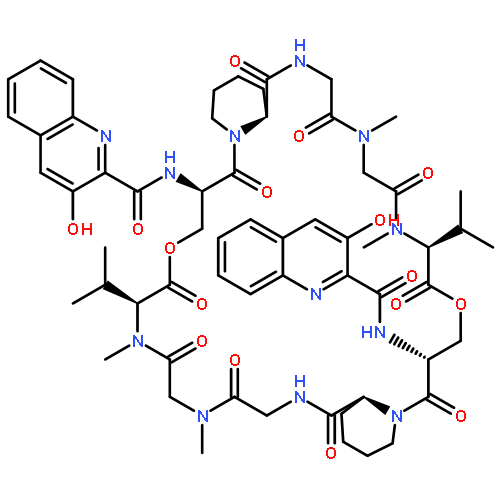
Co-reporter:Akira Katsuyama, Atmika Paudel, Suresh Panthee, Hiroshi Hamamoto, Toru Kawakami, Hironobu Hojo, Fumika Yakushiji, and Satoshi Ichikawa
Organic Letters July 21, 2017 Volume 19(Issue 14) pp:3771-3771
Publication Date(Web):July 11, 2017
DOI:10.1021/acs.orglett.7b01629
The total synthesis of plusbacin A3 (1) has been accomplished using a solvent-dependent diastereodivergent Joullié–Ugi three-component reaction (JU-3CR) as a key step. Two trans-3-hydroxy-l-proline residues were constructed by combining the JU-3CR with a convertible isocyanide strategy. Subsequent peptide coupling and macrolactamization afforded plusbacin A3. Investigating the antibacterial activity of 1 compared with that of its dideoxy analogue revealed that the threo-β-hydroxyaspartic acid residues are essential for antibacterial activity. Notably, there is a low potential for the development of resistance in S. aureus against plusbacin A3.
Co-reporter:S. Kitahata;F. Yakushiji;S. Ichikawa
Chemical Science (2010-Present) 2017 vol. 8(Issue 10) pp:6959-6963
Publication Date(Web):2017/09/25
DOI:10.1039/C7SC02941A
Molecules that have a reactive functional group within a macrocycle represent a class of covalent inhibitor. The relationship between reactivity and affinity for the target is cooperative and complicated. An understanding and characterization of this class of inhibitor are vital for the development of covalent inhibitors as drug candidates. Herein, we describe a systematic analysis of structure–activity relationships using a series of syringolin analogues, which are irreversible covalent inhibitors of proteasomes. We investigate the detailed mechanistic effects of the macrocycles on affinity and reaction rate.
Co-reporter:Shun Kitahata, Takuya Chiba, Takashi Yoshida, Masaki Ri, Shinsuke Iida, Akira Matsuda, and Satoshi Ichikawa
Organic Letters 2016 Volume 18(Issue 9) pp:2312-2315
Publication Date(Web):April 28, 2016
DOI:10.1021/acs.orglett.6b01053
Isosyringolin A, which is an isomer of the proteasome-inhibiting natural product syringolin A, was designed and synthesized to develop analogues that are step economical and synthetically accessible in a practical manner. It was revealed that isosyringolin A exhibited proteasome-inhibitory activity comparable to that of syringolin A and that its derivatization leads to great enhancement in its proteasome inhibitory activity as well as its cytotoxicity against human myeloma cells.
Co-reporter:Akira Katsuyama, Akira Matsuda, and Satoshi Ichikawa
Organic Letters 2016 Volume 18(Issue 11) pp:2552-2555
Publication Date(Web):May 23, 2016
DOI:10.1021/acs.orglett.6b00827
The effect of the solvent on the diastereoselectivity of the Joullié–Ugi three-component reaction (JU-3CR) using an α-substituted five-membered cyclic imine is revisited. The cis and trans isomers were generated in toluene and HFIP, respectively. Hammett analysis of the JU-3CR suggests the presence of two reaction mechanisms.
Co-reporter:Satoshi Ichikawa
The Chemical Record 2016 Volume 16( Issue 3) pp:1106-1115
Publication Date(Web):
DOI:10.1002/tcr.201500247
Abstract
It is important to pursue function-oriented synthesis (FOS), a strategy for the design of less structurally complex targets with comparable or superior activity that can be made in a practical manner, because compared to synthetic drugs, many biologically relevant natural products possess large and complex chemical structures that may restrict chemical modifications in a structure–activity relationship study. In this account, we describe recent efforts to simplify complex nucleoside natural products including caprazamycins. Considering the structure–activity relationship study with several truncated analogues, three types of simplified derivatives, namely, oxazolidine, isoxazolidine, and lactam-fused isoxazolidine-containing uridine derivatives, were designed and efficiently synthesized. These simplified derivatives have exhibited promising antibacterial activities. A significant feature of our studies is the rational and drastic simplification of the molecular architecture of caprazamycins. This study provides a novel strategy for the development of a new type of antibacterial agent effective against drug-resistant bacteria.
Co-reporter:Mayumi Yamaguchi, Akira Matsuda and Satoshi Ichikawa
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 4) pp:1187-1197
Publication Date(Web):14 Nov 2014
DOI:10.1039/C4OB02142H
Simplification of caprazamycins, which are promising antibacterial nucleoside natural products, was conducted by scaffold-hopping of the structurally complex diazepanone moiety to the isoxazolidine scaffold. The designed isoxazolidine-containing uridine derivatives were synthesized by an intramolecular 1,3-dipolar cycloaddition of alkenyl nitrone as a key step. The lactone-fused isoxazolidine intermediate was easily converted to the target compounds by sequential introduction of key substituents upon ring-opening the lactone moiety by nucleophilic substitution and electrophilic capping of the resulting primary alcohol. Several analogues exhibited good activity against H. influenzae ATCC 10211 (MIC 0.25–0.5 μg mL−1) and moderate activity against vancomycin-resistant E. faecalis SR7914 (MIC 4–8 μg mL−1).
Co-reporter:Takeshi Nakaya, Akira Matsuda and Satoshi Ichikawa
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 28) pp:7720-7735
Publication Date(Web):09 Jun 2015
DOI:10.1039/C5OB01037C
The discovery of new antibiotics is critical because the emergence of drug-resistant bacteria has posed a serious public health problem. Caprazamycins, which are nucleoside antibiotics, are a promising lead with a novel mode of action, and we have designed simplified analogues containing a piperidine as a scaffold linking the crucial structural units of caprazamycins. These analogues were step-economically synthesized via a sequential aza-Prins–Ritter reaction. Among the tested compounds, the analogue 7 exhibited good MraY inhibitory activity, antibacterial activity against Gram-positive bacterial strains including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococci, and metabolic stability. The observed cytotoxicity of 7 against HepG2 cells was overcome by modulating the fatty acyl side chain. The knowledge obtained from our structure–activity relationship studies of the caprazamycins will provide further direction toward the design of potent MraY inhibitors.
Co-reporter:Takuya Chiba, Akira Matsuda, Satoshi Ichikawa
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 21) pp:4872-4877
Publication Date(Web):1 November 2015
DOI:10.1016/j.bmcl.2015.06.015
A detailed structure–activity relationship of syringolin A (1), which is a promising antitumor natural product, was described. We previously developed syringolin A analog 2 as a potent proteasome inhibitor by the structure-based drug design of syringolin A. In this Letter, we synthesized a range of analogs of 2, having a different length of the lipophilic chain and substituted aryl group, and their cytotoxicity against human cancer cells was evaluated. It turned out that these modifications greatly affected the cytotoxicity. Further optimization would lead to develop a novel proteasome inhibitor.
Co-reporter:Satoshi Ichikawa, Mayumi Yamaguchi, Lee Shang Hsuan, Yuta Kato, and Akira Matsuda
ACS Infectious Diseases 2015 Volume 1(Issue 4) pp:151
Publication Date(Web):February 19, 2015
DOI:10.1021/id5000376
Carbacaprazamycins, which are chemically stable analogues of caprazamycins, were designed and synthesized. These analogues were active against drug-resistant bacterial pathogens such as methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci, and their activities were comparable to those of the parent caprazamycins. The effect of treatment with carbacaprazamycin on morphological changes in S. aureus indicated that the mode of action was completely different from those of existing peptidoglycan inhibitors.Keywords: antibiotics; caprazamycins; drug resistance; MraY; peptidoglycan
Co-reporter:Katsushi Katayama, Koji Nakagawa, Hiroshi Takeda, Akira Matsuda, and Satoshi Ichikawa
Organic Letters 2014 Volume 16(Issue 2) pp:428-431
Publication Date(Web):December 17, 2013
DOI:10.1021/ol403319m
The total synthesis of sandramycin has been accomplished by using a Staudinger/aza-Wittig/diastereoselective Ugi three-component reaction sequence as a key step to obtain a linear pentadepsipeptide. Subsequent [5 + 5] coupling of the penptapeptide, macrolactamization, and introduction of the quinaldin chromophores afforded sandramycin. Dihydroxy and diacetoxy analogues were also prepared, and the cytotoxic activity of these analogues against a range of human cancer cell lines was evaluated.
Co-reporter:Tetsuya Tanino;Mayumi Yamaguchi;Akira Matsuda
European Journal of Organic Chemistry 2014 Volume 2014( Issue 9) pp:1836-1840
Publication Date(Web):
DOI:10.1002/ejoc.201400140
Abstract
Function-oriented synthesis of a class of liponucleoside antibiotics was investigated through rational simplification guided by previous structure–activity relationship studies of caprazamycins and muraymycins to address the issue associated with their molecular complexity. A lactam-fused isoxazolidine scaffold was designed, and a diverse set of lactam-fused isoxazolidines derivatives were constructed by intramolecular 1,3-dipolar cycloaddition of alkenyl nitrones. Several analogues exhibited moderate activity against a range of Gram-positive drug-resistant bacterial pathogens.
Co-reporter:Yusuke Takeoka, Tetsuya Tanino, Mitsuaki Sekiguchi, Shuji Yonezawa, Masahiro Sakagami, Fumiyo Takahashi, Hiroko Togame, Yoshikazu Tanaka, Hiroshi Takemoto, Satoshi Ichikawa, and Akira Matsuda
ACS Medicinal Chemistry Letters 2014 Volume 5(Issue 5) pp:556-560
Publication Date(Web):March 6, 2014
DOI:10.1021/ml5000096
It is urgent to develop novel anti-Pseudomonas agents that should also be active against multidrug resistant P. aeruginosa. Expanding the antibacterial spectrum of muraymycins toward P. aeruginosa was investigated by the systematic structure–activity relationship study. It was revealed that two functional groups, a lipophilic side chain and a guanidino group, at the accessory moiety of muraymycins were important for the anti-Pseudomonas activity, and analogue 29 exhibited antibacterial activity against a range of P. aeruginosa strains with the minimum inhibitory concentration values of 4–8 μg/mL.Keywords: anti-Pseudomonas; Antibiotics; drug-resistance; MraY; muraymycin; peptidoglycan;
Co-reporter:Takuya Chiba;Hidetaka Hosono;Koji Nakagawa;Masahiro Asaka;Hiroshi Takeda;Akira Matsuda
Angewandte Chemie International Edition 2014 Volume 53( Issue 19) pp:4836-4839
Publication Date(Web):
DOI:10.1002/anie.201402428
Abstract
The development process for syringolin A analogues having improved proteasome inhibitory and antitumor activity is described. The strategy was to first establish a convergent synthesis of syringolin A using a rare intramolecular Ugi three-component reaction in the last stage of the synthesis, so as to gain access toa set of structure-based analogues. The inhibitory activity of chymotrypsin-like activity of 20S proteasome was largely improved by targeting the S3 subsite of the β5 subunit. Cytotoxic activity was also improved by installing the membrane-permeable substituent. These biological properties are comparable to those of bortezomib, a clinically used first-line proteasome inhibitor.
Co-reporter:Takuya Chiba;Hidetaka Hosono;Koji Nakagawa;Masahiro Asaka;Hiroshi Takeda;Akira Matsuda
Angewandte Chemie 2014 Volume 126( Issue 19) pp:4936-4939
Publication Date(Web):
DOI:10.1002/ange.201402428
Abstract
The development process for syringolin A analogues having improved proteasome inhibitory and antitumor activity is described. The strategy was to first establish a convergent synthesis of syringolin A using a rare intramolecular Ugi three-component reaction in the last stage of the synthesis, so as to gain access toa set of structure-based analogues. The inhibitory activity of chymotrypsin-like activity of 20S proteasome was largely improved by targeting the S3 subsite of the β5 subunit. Cytotoxic activity was also improved by installing the membrane-permeable substituent. These biological properties are comparable to those of bortezomib, a clinically used first-line proteasome inhibitor.
Co-reporter:Katsushi Katayama, Takuya Okamura, Takuya Sunadome, Koji Nakagawa, Hiroshi Takeda, Motoo Shiro, Akira Matsuda, and Satoshi Ichikawa
The Journal of Organic Chemistry 2014 Volume 79(Issue 6) pp:2580-2590
Publication Date(Web):February 20, 2014
DOI:10.1021/jo500039d
The second-generation total synthesis of quinaldopeptin (1) was established via a Staudinger/aza-Wittig/diastereoselective Ugi three-component reaction sequence and a racemization-free [5 + 5] coupling and macrolactamization. A single-crystal X-ray structure of the chromophore analogue 26 confirmed the structural and stereochemical assignments of the macrocycle. Synthetic 1 successfully unwound supercoiled DNA to form a relaxed DNA in a dose-dependent manner, the binding affinity of 1 to four dsODNs was within a similar range (Kb = 1.45–2.53 × 107 M–1), and the sequence selectivity was subtle. It was suggested that 1 possesses biological behaviors similar to those of sandramycin (2) in terms of cytotoxic activity against human cancer cell lines (IC50 = 3.2–12 nM) and HIF-1 inhibitory activity.
Co-reporter:Satoshi Ichikawa, Hideaki Ueno, Takuya Sunadome, Kousuke Sato, and Akira Matsuda
Organic Letters 2013 Volume 15(Issue 3) pp:694-697
Publication Date(Web):January 22, 2013
DOI:10.1021/ol400001w
Triazole-cross-linked oligodeoxynucleotides were synthesized using the Cu(I) catalyzed alkyne–azide cycloaddition with tris(azidoethyl)amine hydrochloride and oligodeoxynucleotides possessing N-3-(propargyl)thymidine at both the 3′- and 5′-termini. Further installation of a functional molecule to the dumbbell oligodeoxynucleotides was achieved by utilizing the remaining azide group.
Co-reporter:Satoshi Ichikawa, Nana Tatebayashi, and Akira Matsuda
The Journal of Organic Chemistry 2013 Volume 78(Issue 23) pp:12065-12075
Publication Date(Web):October 15, 2013
DOI:10.1021/jo4020672
Indolocarbazole natural products are known to possess a variety of biological activities that hold promise as cancer chemotherapeutic agents. We newly designed C-glycosyl pyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione derivatives 7 and 8, which are natural-product-like scaffolds. Compounds 7 and 8 were stereoselectively and efficiently synthesized using β-selective C-allylation, Heck reaction, and thermal 6π-electron cyclization/oxidative aromatization. Their potential as Chk1 inhibitors was investigated, and 7 and 8 exhibited an inhibitory activity with IC50 values of 0.5–9.5 μM, which is good activity for scaffolds. The key intermediate 23 was obtained by five steps from d-ribose in 33% overall yield by this synthetic route, which would enable us to prepare a range of analogues in order to investigate further structure–activity relationship studies in the optimization process.
Co-reporter:Satoshi Ichikawa, Takuya Okamura, and Akira Matsuda
The Journal of Organic Chemistry 2013 Volume 78(Issue 24) pp:12662-12670
Publication Date(Web):November 15, 2013
DOI:10.1021/jo402267r
The first total synthesis of quinaldopeptin (1) was accomplished. Our approach to the synthesis of 1 includes the solid-phase peptide synthesis of the linear decapeptide 4 followed by macrocyclization and introduction of the quinoline chromophores 2 at a late stage of the synthesis. As for the preparation of 4, a fragment coupling approach was applied considering the C2 symmetrical structure of 1. Chromophore analogues 22 and 23 and desmethyl analogue 27 were also prepared in a manner similar to the synthesis of 1. Synthetic 1 exhibits a strong cytotoxicity with the IC50 value of 3.2 nM. On the other hand, the activity of 23 and 27 was largely reduced.
Co-reporter:Kazuya Okamoto, Masahiro Sakagami, Fei Feng, Fumiyo Takahashi, Kouichi Uotani, Hiroko Togame, Hiroshi Takemoto, Satoshi Ichikawa, Akira Matsuda
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4810-4815
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.050
The second-generation synthesis of 3′-hydroxypacidamycin D (2) has been accomplished via an Ugi-four component reaction at a late stage of the synthesis. This approach provided ready access to a range of analogues including diastereomers of the diaminobutylic acid residue and hybrid-type analogues of mureidomycins. Biological evaluations of these analogues indicated that the stereochemistry at the diaminobutylic acid residue has a crucial impact on both the MraY biochemical inhibition and whole-cell antibacterial activity.
Co-reporter:Kazuya Okamoto, Masahiro Sakagami, Fei Feng, Hiroko Togame, Hiroshi Takemoto, Satoshi Ichikawa, and Akira Matsuda
The Journal of Organic Chemistry 2012 Volume 77(Issue 3) pp:1367-1377
Publication Date(Web):December 22, 2011
DOI:10.1021/jo202159q
Full details of the total synthesis of pacidamycin D (4) and its 3′-hydroxy analogue 32 are described. The chemically labile Z-oxyacyl enamide moiety is the most challenging chemical structure found in uridylpeptide natural products. Key elements of our approach to the synthesis of 4 include the efficient and stereocontrolled construction of the Z-oxyvinyl halides 6 and 7 and their copper-catalyzed cross-coupling with the tetrapeptide carboxamide 5, a thermally unstable compound containing a number of potentially reactive functional groups. This synthetic route also allowed us to easily prepare 3′-hydroxy analogue 32. The assemblage by cross-coupling of the Z-oxyvinyl halide 6 and the carboxamide 5 at a late stage of the synthesis provided ready access to a range of uridylpeptide antibiotics and their analogues, despite their inherent labile nature with potential epimerization, simply by altering the tetrapeptide moiety.
Co-reporter:Tetsuya Tanino ; Bayan Al-Dabbagh ; Dominique Mengin-Lecreulx ; Ahmed Bouhss ; Hiroshi Oyama ; Satoshi Ichikawa ;Akira Matsuda
Journal of Medicinal Chemistry 2011 Volume 54(Issue 24) pp:8421-8439
Publication Date(Web):November 15, 2011
DOI:10.1021/jm200906r
The systematic structure–activity relationship (SAR) of the muraymycins (MRYs) using an Ugi four-component reaction (U4CR) was investigated. The impact of the lipophilic substituent on antibacterial activity was significant, and the analogues 8 and 9 having a lipophilic side chain exhibited good activity against a range of Gram-positive bacterial pathogens, including MRSA and VRE. Further investigation of compounds 8 and 9 revealed these analogues to be selective inhibitors of the MraY transferase and nontoxic to HepG2 cells. The SAR of the accessory urea–peptide moiety indicated that it could be simplified. Our SAR study of the MRYs suggests a probable mechanism for inhibition of the MraY, where the inner moiety of the urea–dipeptide motif interacts with the carbohydrate recognition domain in the cytoplasmic loop 5. The predicted binding model would provide further direction toward the design of potent MraY inhibitors. This study has set the stage for the generation of novel antibacterial “lead” compounds based on MRYs.
Co-reporter:Kazuhiro Muranaka, Satoshi Ichikawa, and Akira Matsuda
The Journal of Organic Chemistry 2011 Volume 76(Issue 22) pp:9278-9293
Publication Date(Web):October 12, 2011
DOI:10.1021/jo201495w
The new carboxamide protecting group, 4-(tert-butyldimethylsiloxy)-2-methoxybenzyl (SiMB), has been developed. While this SiMB group can be removed using mild basic desilylation methods, it can also be deprotected under strongly acidic or oxidative conditions. An application of this group to simple carboxamide groups, as well as to more complex and acid-sensitive adenosine derivatives containing a cyclophane scaffold, was also demonstrated.
Co-reporter:Kensuke Ii ; Satoshi Ichikawa ; Bayan Al-Dabbagh ; Ahmed Bouhss ;Akira Matsuda
Journal of Medicinal Chemistry 2010 Volume 53(Issue 9) pp:3793-3813
Publication Date(Web):April 20, 2010
DOI:10.1021/jm100243n
The rational simplification of the caprazamycin (CPZ) class of nucleoside natural products was carried out to address their molecular complexity. First, analogues 6−8, where the diazepanone ring of the CPZ was removed and a lipophilic side chain was attached to either the C-7′ or N6′ atom, were used to investigate the conformation−activity relationship. On the basis of this relationship, we designed the oxazolidine-containing uridine derivatives 18−21 by restricting the conformation of 6−8. As a result, the tBu ester derivatives 20 were found to be the most active against a range of bacterial strains containing VRE with a potency similar to that of the parent CPZs. This study provides a novel strategy for the development of a new type of antibacterial agent effective against drug-resistant bacteria.
Co-reporter:Tetsuya Tanino, Satoshi Ichikawa, Bayan Al-Dabbagh, Ahmed Bouhss, Hiroshi Oyama and Akira Matsuda
ACS Medicinal Chemistry Letters 2010 Volume 1(Issue 6) pp:258
Publication Date(Web):June 2, 2010
DOI:10.1021/ml100057z
Muraymycin analogues with a lipophilic substituent were synthesized using an Ugi four-component assemblage. This approach provides ready access to a range of analogues simply by altering the aldehyde component. The impact of the lipophilic substituent on the antibacterial activity was very large, and analogues 7b−e and 8b−e exhibited good activity against a range of Gram-positive bacterial pathogens including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus faecium. This study also showed that the accessory urea-dipeptide motif contributes to MraY inhibitory and antibacterial activity. The knowledge obtained from our structure−activity relationship study of muraymycins provides further direction toward the design of potent MraY inhibitors. This study has set the stage for the generation of novel antibacterial “lead” compounds based on muraymycins.Keywords (keywords): Antibiotics; drug resistance; MraY; muraymycin; peptidoglycan; Ugi four-component reaction
Co-reporter:Yuki Sako, Satoshi Ichikawa, Akiko Osada, Akira Matsuda
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 22) pp:7878-7889
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmc.2010.09.042
The structure–activity relationship (SAR) of 5-substituted pyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione derivatives 5 was investigated for their potential as Chk1 inhibitors for possible chemo- and radio-potentiators in anticancer chemotherapies. In silico virtual screening helped to optimize the substituent on the phenyl ring, and led to identification of the m-carbamoyl group among the 117 analogues tested. Further optimization studies focusing on the docking model of 15 in the active site of Chk1 revealed that 32b (IC50 = 2.8 nM) was a more potent inhibitor than UNC-01.Potent inhibitors of Check point kinase 1 have been developed by a structure-based drug design.
Co-reporter:Hironori Sekiguchi, Kazuhiro Muranaka, Akiko Osada, Satoshi Ichikawa, Akira Matsuda
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 15) pp:5732-5737
Publication Date(Web):1 August 2010
DOI:10.1016/j.bmc.2010.05.075
The PU-H58-dimers 13a–15b were efficiently synthesized and their biological properties were evaluated. The copper-catalyzed alkyne azide coupling was effective in simultaneously linking three components via a triazole formation to afford the target dimers. These synthesized dimers exhibited binding affinity to the N-terminal domain of Hsp90, cytotoxicity, and client degradation activity although these activities were comparative or weak comparable with that of the parent compound.The PU-H58-dimers were designed as potential Hsp90 inhibitors and efficiently synthesized by the copper-catalyzed alkyne azide coupling (CuAAC) and their biological properties including binding affinity to the N-terminal domain of Hsp90, cytotoxicity, and client degradation activity were evaluated.
Co-reporter:Tetsuya Tanino, Satoshi Ichikawa, Motoo Shiro and Akira Matsuda
The Journal of Organic Chemistry 2010 Volume 75(Issue 5) pp:1366-1377
Publication Date(Web):February 9, 2010
DOI:10.1021/jo9027193
Full details of the first total synthesis of (−)-muraymycin (MRY) D2 and its epimer, the antibacterial nucleoside natural product, are described. Key strategic elements of the approach include the preparation of the urea dipeptide moiety found in the muraymycins containing an l-epi-capreomycidine via a nitrene C−H insertion of the sulfamate 10 and the fully protected muraymycin skeleton at a late stage by an Ugi four-component reaction. Thus, the nitrene C−H insertion of the sulfamate 10 with 10 mol % of Rh2(esp)2 catalyst gave the cyclic sulfamates 11a and 11b in 47% yield (11a:11b = 1:2.0). Construction of the cyclic guanidine skeleton was effected through the HgBr2-promoted cyclization of 42 followed by desulfonylation upon acetolysis of the oxathiazinane ring to give 43 in good yield. The amine obtained by selective removal of the Cbz group of the alcohol 44 was reacted with MeSC(═O)-l-Val-O-t-Bu (38) to provide 45, which was oxidized to the carboxylic acid 46. Reaction of 46, isonitrile 51, isovaleraldehyde, and 2,4-dimethoxybenzylamine furnished the desired Ugi products, the final deprotection of which successfully afforded (−)-MRY D2 and epi-MRY D2 (53) after HPLC separation of the diastereomers. This approach would afford ready access to a range of analogues simply by altering each component.
Co-reporter:Kazuhiro Muranaka, Akiko Sano, Satoshi Ichikawa, Akira Matsuda
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 11) pp:5862-5870
Publication Date(Web):1 June 2008
DOI:10.1016/j.bmc.2008.04.070
Structure-based drug design was used to systematically synthesize PU3-dimers. The cytotoxicity of PU3 dimers 6 against breast cancer cell lines was evaluated, and their potency increased as the length of the bridging linker increased. Among the compounds tested, 6e with a C-20 linker was the most potent and exhibited a 20- to 30-fold increase in activity compared with that of the parent compound 5. Western blot analyses of the cell lysates treated with 6c revealed that 6c resulted in the concentration-dependent degradation of the Hsp90 client protein Her2, which is consistent with other Hsp90 inhibitors.Structure-based drug design was used to systematically synthesize PU3-dimers. Their cytotoxic potency increased as the length of the bridging linker increased.
Co-reporter:Tetsuya Tanino ; Satoshi Ichikawa ;Akira Matsuda
Organic Letters () pp:
Publication Date(Web):July 7, 2011
DOI:10.1021/ol201527k
The l-epi-capreomycidine (Cpm) derivatives were efficiently and stereoselectively synthesized via nitrene C–H insertion starting from a readily available d-Tyr. Design of a substrate that takes into account hydrogen bonding is a critical feature in order to achieve high selectivity. Our synthetic strategy could be a new access to epi-Cpm and its derivatives, which are found in several biologically active natural products.
Co-reporter:Mayumi Yamaguchi, Akira Matsuda and Satoshi Ichikawa
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 4) pp:NaN1197-1197
Publication Date(Web):2014/11/14
DOI:10.1039/C4OB02142H
Simplification of caprazamycins, which are promising antibacterial nucleoside natural products, was conducted by scaffold-hopping of the structurally complex diazepanone moiety to the isoxazolidine scaffold. The designed isoxazolidine-containing uridine derivatives were synthesized by an intramolecular 1,3-dipolar cycloaddition of alkenyl nitrone as a key step. The lactone-fused isoxazolidine intermediate was easily converted to the target compounds by sequential introduction of key substituents upon ring-opening the lactone moiety by nucleophilic substitution and electrophilic capping of the resulting primary alcohol. Several analogues exhibited good activity against H. influenzae ATCC 10211 (MIC 0.25–0.5 μg mL−1) and moderate activity against vancomycin-resistant E. faecalis SR7914 (MIC 4–8 μg mL−1).
Co-reporter:Takeshi Nakaya, Akira Matsuda and Satoshi Ichikawa
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 28) pp:NaN7735-7735
Publication Date(Web):2015/06/09
DOI:10.1039/C5OB01037C
The discovery of new antibiotics is critical because the emergence of drug-resistant bacteria has posed a serious public health problem. Caprazamycins, which are nucleoside antibiotics, are a promising lead with a novel mode of action, and we have designed simplified analogues containing a piperidine as a scaffold linking the crucial structural units of caprazamycins. These analogues were step-economically synthesized via a sequential aza-Prins–Ritter reaction. Among the tested compounds, the analogue 7 exhibited good MraY inhibitory activity, antibacterial activity against Gram-positive bacterial strains including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococci, and metabolic stability. The observed cytotoxicity of 7 against HepG2 cells was overcome by modulating the fatty acyl side chain. The knowledge obtained from our structure–activity relationship studies of the caprazamycins will provide further direction toward the design of potent MraY inhibitors.

![2,4-Dodecadienamide,N-[(1S,2R)-2-hydroxy-1-[[[(3E,5S,8S,10S)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl]amino]carbonyl]propyl]-,(2E,4E)-](http://img.cochemist.com/ccimg/108400/108351-50-4.png)
![2,4-Dodecadienamide,N-[(1S,2R)-2-hydroxy-1-[[[(3E,5S,8S,10S)-10-hydroxy-5-methyl-2,7-dioxo-1,6-diazacyclododec-3-en-8-yl]amino]carbonyl]propyl]-,(2E,4E)-](http://img.cochemist.com/ccimg/108400/108351-50-4_b.png)


![SU 9516;3-[1-(3H-IMIDAZOL-4-YL)-METH-(Z)-YLIDENE]-5-METHOXY-1,3-DIHYDRO-INDOL-2-ONE](http://img.cochemist.com/ccimg/377100/377090-84-1.png)
![SU 9516;3-[1-(3H-IMIDAZOL-4-YL)-METH-(Z)-YLIDENE]-5-METHOXY-1,3-DIHYDRO-INDOL-2-ONE](http://img.cochemist.com/ccimg/377100/377090-84-1_b.png)
![2-[METHYL-[2-[(2-METHYLPROPAN-2-YL)OXYCARBONYLAMINO]ACETYL]AMINO]ACETIC ACID](http://img.cochemist.com/ccimg/133500/133498-97-2.png)
![2-[METHYL-[2-[(2-METHYLPROPAN-2-YL)OXYCARBONYLAMINO]ACETYL]AMINO]ACETIC ACID](http://img.cochemist.com/ccimg/133500/133498-97-2_b.png)








![D-Serine, N-[(phenylmethoxy)carbonyl]-, 2,2,2-trichloroethyl ester](http://img.cochemist.com/ccimg/78800/78700-33-1.png)
![D-Serine, N-[(phenylmethoxy)carbonyl]-, 2,2,2-trichloroethyl ester](http://img.cochemist.com/ccimg/78800/78700-33-1_b.png)