Co-reporter:Shorouk Dannoon; Tanushree Ganguly; Hendry Cahaya; Jonathan J. Geruntho; Matthew S. Galliher; Sophia K. Beyer; Cindy J. Choy; Mark R. Hopkins; Melanie Regan; Joseph E. Blecha; Lubica Skultetyova; Christopher R. Drake; Salma Jivan; Cyril Barinka; Ella F. Jones; Clifford E. Berkman;Henry F. VanBrocklin
Journal of Medicinal Chemistry 2016 Volume 59(Issue 12) pp:5684-5694
Publication Date(Web):May 26, 2016
DOI:10.1021/acs.jmedchem.5b01850
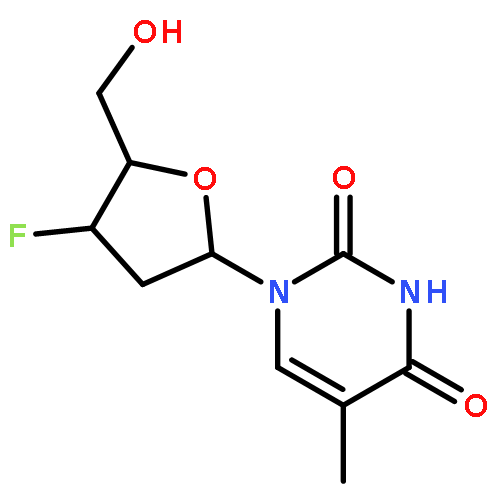
A series of phosphoramidate-based prostate specific membrane antigen (PSMA) inhibitors of increasing lipophilicity were synthesized (4, 5, and 6), and their fluorine-18 analogs were evaluated for use as positron emission tomography (PET) imaging agents for prostate cancer. To gain insight into their modes of binding, they were also cocrystallized with the extracellular domain of PSMA. All analogs exhibited irreversible binding to PSMA with IC50 values ranging from 0.4 to 1.3 nM. In vitro assays showed binding and rapid internalization (80–95%, 2 h) of the radiolabeled ligands in PSMA(+) cells. In vivo distribution demonstrated significant uptake in CWR22Rv1 (PSMA(+)) tumor, with tumor to blood ratios of 25.6:1, 63.6:1, and 69.6:1 for [18F]4, [18F]5, and [18F]6, respectively, at 2 h postinjection. Installation of aminohexanoic acid (AH) linkers in the phosphoramidate scaffold improved their PSMA binding and inhibition and was critical for achieving suitable in vivo imaging properties, positioning [18F]5 and [18F]6 as favorable candidates for future prostate cancer imaging clinical trials.
Co-reporter:Aaron M. LeBeau;Minhee Lee;Stephanie T. Murphy;Byron C. Hann;Robert S. Warren;Romelyn Delos Santos;John Kurhanewicz;Samir M. Hanash;Henry F. VanBrocklin;Charles S. Craik;
Proceedings of the National Academy of Sciences 2013 110(1) pp:93-98
Publication Date(Web):December 17, 2012
DOI:10.1073/pnas.1218694110
Proteases responsible for the increased peritumoral proteolysis associated with cancer represent functional biomarkers for
monitoring tumorigenesis. One attractive extracellular biomarker is the transmembrane serine protease matriptase. Found on
the surface of epithelial cells, the activity of matriptase is regulated by its cognate inhibitor hepatocyte growth factor
activator inhibitor-1 (HAI-1). Quantitative mass spectrometry allowed us to show that, in selected cancers, HAI-1 expression
decreases, leading to active matriptase. A preclinical probe specific for the measurement of emergent active matriptase was
developed. Using an active-site–specific, recombinant human antibody for matriptase, we found that the selective targeting
of active matriptase can be used to visualize the tumorigenic epithelium. Live-cell fluorescence imaging validated the selectivity
of the antibody in vitro by showing that the probe localized only to cancer cell lines with active matriptase on the surface.
Immunofluorescence with the antibody documented significant levels of active matriptase in 68% of primary and metastatic colon
cancer sections from tissue microarrays. Labeling of the active form of matriptase in vivo was measured in human colon cancer
xenografts and in a patient-derived xenograft model using near-infrared and single-photon emission computed tomography imaging.
Tumor uptake of the radiolabeled antibody, 111In-A11, by active matriptase was high in xenografts (28% injected dose per gram) and was blocked in vivo by the addition of
a matriptase-specific variant of ecotin. These findings suggest, through a HAI-1–dependent mechanism, that emergent active
matriptase is a functional biomarker of the transformed epithelium and that its proteolytic activity can be exploited to noninvasively
evaluate tumorigenesis in vivo.
Co-reporter:Neil Vasdev, Peter N. Dorff, James P. O’Neil, Frederick T. Chin, Stephen Hanrahan, Henry F. VanBrocklin
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 9) pp:2959-2965
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmc.2011.03.032
Epidermal growth factor receptors (EGFR), upregulated in many tumor types, have been a target for therapeutic development and molecular imaging. The objective of this study was to evaluate the distribution and metabolic characteristics of fluorine-18 labeled anilinoquinazolines as potential imaging agents for EGFR tyrosine kinase expression. Fluorine-18 labeled fluoronitrobenzenes were prepared by reaction of potassium cryptand [18F]fluoride with 1,2- and 1,4-dinitrobenzenes, and 3-nitro-N,N,N-trimethylanilinium triflate in 5 min. Decay-corrected radiochemical yields of [18F]fluoride incorporation into the nitro-aromatic compounds were 81 ± 2%, 44 ± 4% and 77 ± 5% (n = 3–5) for the 2-, 3- and 4-fluoro isomers, respectively. Sodium borohydride reduction to the corresponding [18F]fluoroanilines was achieved with greater than 80% conversion in 5 min. Coupling of [18F]fluoroaniline-hydrochlorides to 6,7-dimethoxy-4-chloro-quinazoline gave the corresponding 6,7-dimethoxy-4-(2-, 3- and 4-[18F]fluoroanilino)quinazolines in 31 ± 5%, 17 ± 2% and 55 ± 2% radiochemical yield, respectively, while coupling to the 6,7-diethoxy-4-chloro-quinazoline produced 6,7-diethoxy-4-(2-, 3- and 4-[18F]fluoroanilino)quinazolines in 19 ± 6%, 9 ± 3% and 36 ± 6% radiochemical yield, respectively, in 90 min to end of synthesis from [18F]fluoride. Biodistribution of 2- and 4-[18F]fluoroanilinoquinazolines was conducted in tumor-bearing mice (MDA-MB-435 and MDA-MB-468 xenografts). Low tumor uptake (<1% injected dose per gram (ID/g) of tissue up to 3 h postinjection of the radiotracers) was observed. High bone uptake (5–15% ID/g) was noted with the 4-[18F]fluoroanilinoquinazolines. The metabolic stabilities of radiolabeled quinazolines were further evaluated by incubation with human female cryopreserved isolated hepatocytes. Rapid degeneration of the 4-fluoro-substituted compounds to baseline polar metabolites was observed by radio-TLC, whereas, the 2- and 3-[18F]fluoroaniline derivatives were significantly more stable, up to 2 h, corroborating the in vivo biodistribution studies. para-Substituted [18F]fluoroanilines, a common structural motif in radiopharmaceuticals, are highly susceptible to metabolic degradation.




![Octanoic acid, 8-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/144700/144682-03-1.png)
![Octanoic acid, 8-[[(4-methylphenyl)sulfonyl]oxy]-, ethyl ester](http://img.cochemist.com/ccimg/144700/144682-03-1_b.png)


![L-Norvaline, N-[(1,1-dimethylethoxy)carbonyl]-5-hydroxy-, phenylmethyl ester](/data/chemimg/3760100/91229-97-9.png)
![L-Norvaline, N-[(1,1-dimethylethoxy)carbonyl]-5-hydroxy-, phenylmethyl ester](/data/chemimg/3760100/91229-97-9_b.png)


![CARBAMIC ACID, [6-[(2,5-DIOXO-1-PYRROLIDINYL)OXY]-6-OXOHEXYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/53800/53733-98-5.png)
![CARBAMIC ACID, [6-[(2,5-DIOXO-1-PYRROLIDINYL)OXY]-6-OXOHEXYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/53800/53733-98-5_b.png)