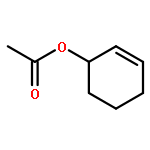
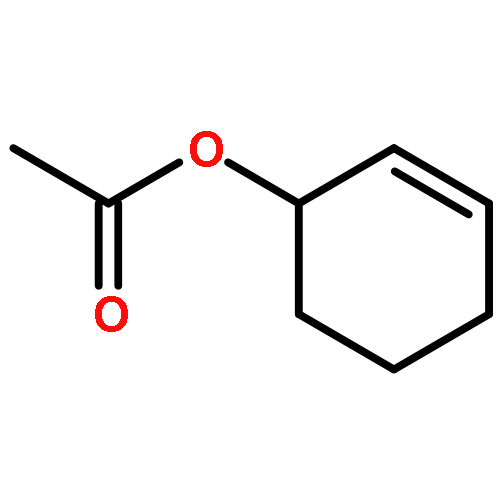
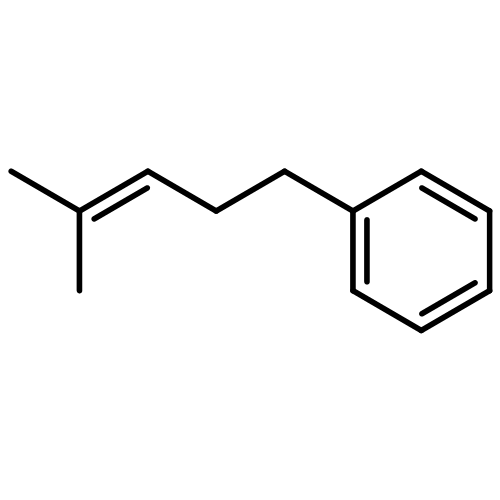
Co-reporter:Chi Chen ; Thomas R. Dugan ; William W. Brennessel ; Daniel J. Weix ;Patrick L. Holland
Journal of the American Chemical Society 2014 Volume 136(Issue 3) pp:945-955
Publication Date(Web):January 3, 2014
DOI:10.1021/ja408238n
The isomerization of simple terminal alkenes to internal isomers with Z-stereochemistry is rare, because the more stable E-isomers are typically formed. We show here that cobalt(II) catalysts supported by bulky β-diketiminate ligands have the appropriate kinetic selectivity to catalyze the isomerization of some simple 1-alkenes specifically to the 2-alkene as the less stable Z-isomer. The catalysis proceeds via an “alkyl” mechanism, with a three-coordinate cobalt(II) alkyl complex as the resting state. β-Hydride elimination and [1,2]-insertion steps are both rapid, as shown by isotopic labeling experiments. A steric model explains the selectivity through a square-planar geometry at cobalt(II) in the transition state for β-hydride elimination. The catalyst works not only with simple alkenes, but also with homoallyl silanes, ketals, and silyl ethers. Isolation of cobalt(I) or cobalt(II) products from reactions with poor substrates suggests that the key catalyst decomposition pathways are bimolecular, and lowering the catalyst concentration often improves the selectivity. In addition to a potentially useful, selective transformation, these studies provide a mechanistic understanding for catalytic alkene isomerization by high-spin cobalt complexes, and demonstrate the effectiveness of steric bulk in controlling the stereoselectivity of alkene formation.
.jpg)




![6H-Dibenzo[b,d]pyran-6-one, 8-chloro-](http://img.cochemist.com/ccimg/742100/742058-81-7.png)
![6H-Dibenzo[b,d]pyran-6-one, 8-chloro-](http://img.cochemist.com/ccimg/742100/742058-81-7_b.png)


![[1,1'-Biphenyl]-2-carboxylic acid, 4-acetyl-, methyl ester](http://img.cochemist.com/ccimg/537800/537715-93-8.png)
![[1,1'-Biphenyl]-2-carboxylic acid, 4-acetyl-, methyl ester](http://img.cochemist.com/ccimg/537800/537715-93-8_b.png)


![Benzene, 1-[(1E)-5-chloro-1-pentenyl]-4-methoxy-](http://img.cochemist.com/ccimg/485400/485397-84-0.png)
![Benzene, 1-[(1E)-5-chloro-1-pentenyl]-4-methoxy-](http://img.cochemist.com/ccimg/485400/485397-84-0_b.png)












![3-Tosyl-6-oxa-3-azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/159600/159555-66-5.png)
![3-Tosyl-6-oxa-3-azabicyclo[3.1.0]hexane](http://img.cochemist.com/ccimg/159600/159555-66-5_b.png)
![Carbamic acid, [phenyl(phenylsulfonyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/155400/155396-71-7.png)
![Carbamic acid, [phenyl(phenylsulfonyl)methyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/155400/155396-71-7_b.png)












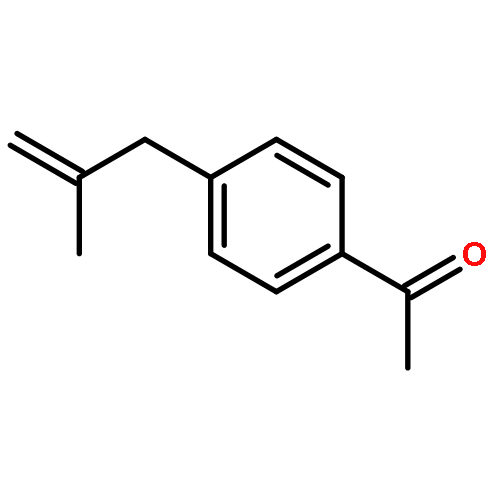
![1-[4-(3-PHENYLPROPYL)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/113200/113138-08-2.png)
![1-[4-(3-PHENYLPROPYL)PHENYL]ETHANONE](http://img.cochemist.com/ccimg/113200/113138-08-2_b.png)




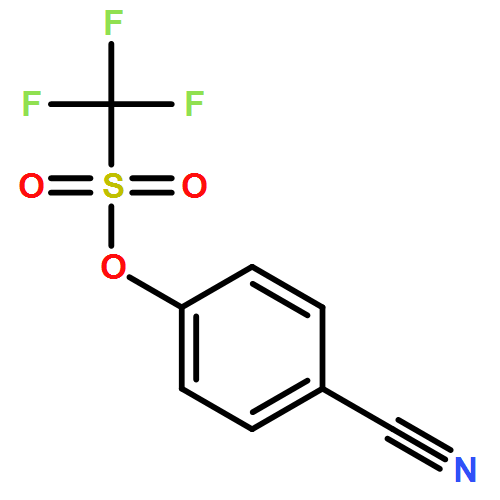
![Benzoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](/data/chemimg/118600/107658-28-6.png)
![Benzoic acid, 3-[[(trifluoromethyl)sulfonyl]oxy]-, methyl ester](/data/chemimg/118600/107658-28-6_b.png)



![1H-INDOLE, 5-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/96600/96546-77-9.png)
![1H-INDOLE, 5-BROMO-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/96600/96546-77-9_b.png)
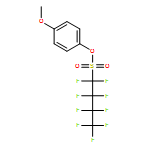





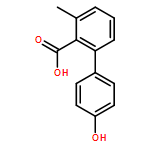
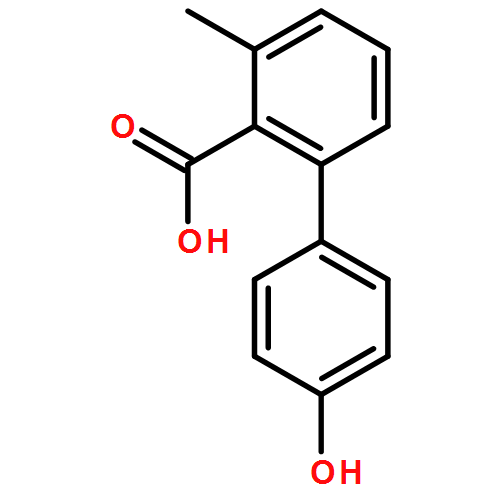
![3-Methyl-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/92300/92254-02-9.png)
![3-Methyl-[1,1'-biphenyl]-2-carboxylic acid](http://img.cochemist.com/ccimg/92300/92254-02-9_b.png)








![3-BENZYL-6-OXA-3-AZABICYCLO[3.1.0]HEXANE](http://img.cochemist.com/ccimg/75400/75390-09-9.png)
![3-BENZYL-6-OXA-3-AZABICYCLO[3.1.0]HEXANE](http://img.cochemist.com/ccimg/75400/75390-09-9_b.png)




![Benzene, 1-[(1Z)-2-bromoethenyl]-4-chloro-](http://img.cochemist.com/ccimg/66500/66482-29-9.png)
![Benzene, 1-[(1Z)-2-bromoethenyl]-4-chloro-](http://img.cochemist.com/ccimg/66500/66482-29-9_b.png)
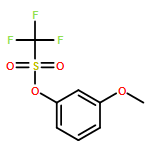
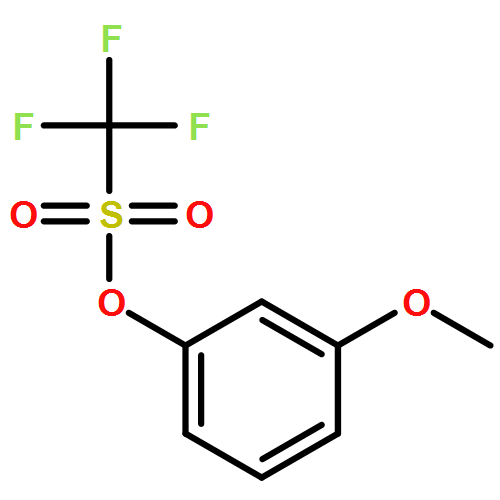
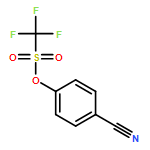



![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6.png)
![Dichloro[bis(1,3-diphenylphosphino)propane]palladium(II)](http://img.cochemist.com/ccimg/59900/59831-02-6_b.png)
![[1,1'-Biphenyl]-4-carbonitrile, 4'-acetyl-](/data/chemimg/1121000/59211-64-2.png)
![[1,1'-Biphenyl]-4-carbonitrile, 4'-acetyl-](/data/chemimg/1121000/59211-64-2_b.png)







![Benzene,[(1E)-3-(trimethylsilyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/40600/40595-34-4.png)
![Benzene,[(1E)-3-(trimethylsilyl)-1-propen-1-yl]-](http://img.cochemist.com/ccimg/40600/40595-34-4_b.png)







![Benzene, [(1Z)-2-bromo-1-propenyl]-](http://img.cochemist.com/ccimg/21500/21453-89-4.png)
![Benzene, [(1Z)-2-bromo-1-propenyl]-](http://img.cochemist.com/ccimg/21500/21453-89-4_b.png)


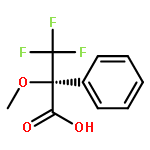
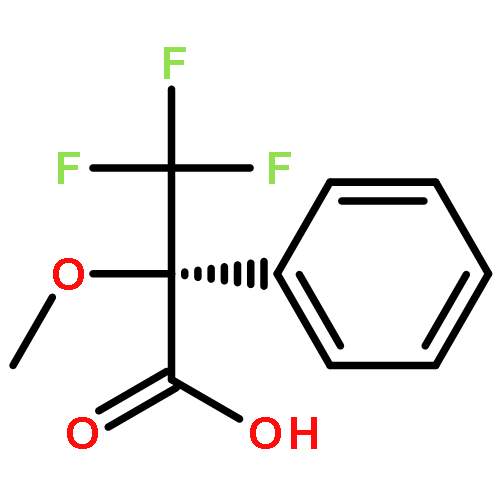


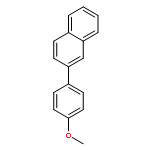
![[1,1'-Biphenyl]-4-amine, 4'-methoxy-N,N-dimethyl-](/data/chemimg/1470800/18158-44-6.png)
![[1,1'-Biphenyl]-4-amine, 4'-methoxy-N,N-dimethyl-](/data/chemimg/1470800/18158-44-6_b.png)





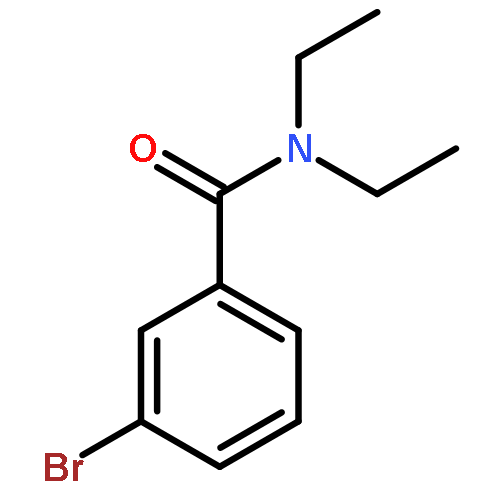
![[1,1'-Biphenyl]-3-carboxylic acid, 4'-fluoro-, methyl ester](http://img.cochemist.com/ccimg/10600/10540-38-2.png)
![[1,1'-Biphenyl]-3-carboxylic acid, 4'-fluoro-, methyl ester](http://img.cochemist.com/ccimg/10600/10540-38-2_b.png)


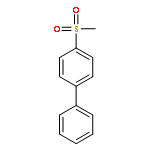

![7-oxabicyclo[4.1.0]hept-3-ene](http://img.cochemist.com/ccimg/6300/6253-27-6.png)
![7-oxabicyclo[4.1.0]hept-3-ene](http://img.cochemist.com/ccimg/6300/6253-27-6_b.png)

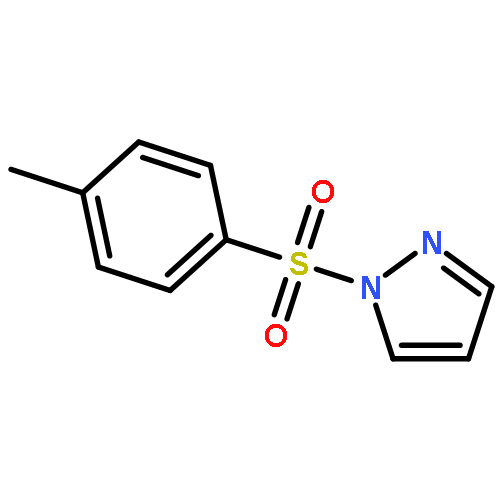
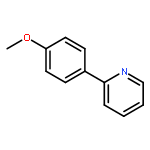
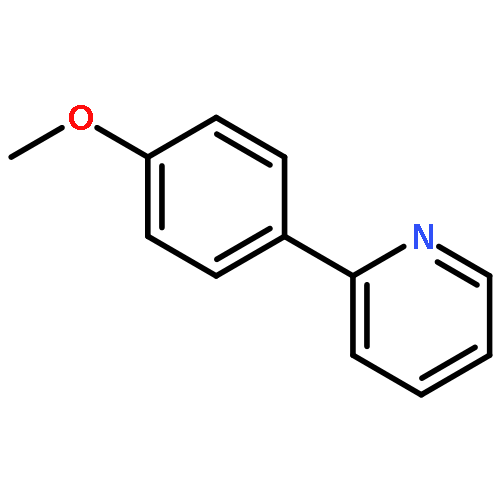
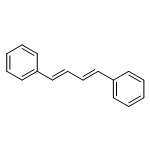








![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0.png)
![Naphth[2,3-b]oxirene,1a,2,7,7a-tetrahydro-](http://img.cochemist.com/ccimg/2500/2461-35-0_b.png)




![6H-Dibenzo[b,d]pyran-6-one](http://img.cochemist.com/ccimg/2100/2005-10-9.png)
![6H-Dibenzo[b,d]pyran-6-one](http://img.cochemist.com/ccimg/2100/2005-10-9_b.png)









![Benzene, [(1Z)-2-bromoethenyl]-](/data/chemimg/1121700/588-73-8.png)
![Benzene, [(1Z)-2-bromoethenyl]-](/data/chemimg/1121700/588-73-8_b.png)


![Benzoic acid, 4-[(2E)-3-phenyl-2-propen-1-yl]-, methyl ester](/data/chemimg/124300/946085-12-7.png)
![Benzoic acid, 4-[(2E)-3-phenyl-2-propen-1-yl]-, methyl ester](/data/chemimg/124300/946085-12-7_b.png)



































![Silane, (1,1-dimethylethyl)[(4-iodophenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/147300/147283-96-3.png)
![Silane, (1,1-dimethylethyl)[(4-iodophenyl)methoxy]dimethyl-](http://img.cochemist.com/ccimg/147300/147283-96-3_b.png)
















![Benzene,[3-[(trimethylsilyl)oxy]-2-cyclohexen-1-yl]-](http://img.cochemist.com/ccimg/108700/108643-81-8.png)
![Benzene,[3-[(trimethylsilyl)oxy]-2-cyclohexen-1-yl]-](http://img.cochemist.com/ccimg/108700/108643-81-8_b.png)












![Phenol, 4-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-](http://img.cochemist.com/ccimg/96100/96013-76-2.png)
![Phenol, 4-[2-[[(1,1-dimethylethyl)dimethylsilyl]oxy]ethyl]-](http://img.cochemist.com/ccimg/96100/96013-76-2_b.png)
![[Bi-1-cyclohexen-1-yl]-6,6'-dione](/data/chemimg/2072800/95251-70-0.png)
![[Bi-1-cyclohexen-1-yl]-6,6'-dione](/data/chemimg/2072800/95251-70-0_b.png)

















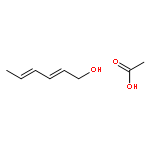
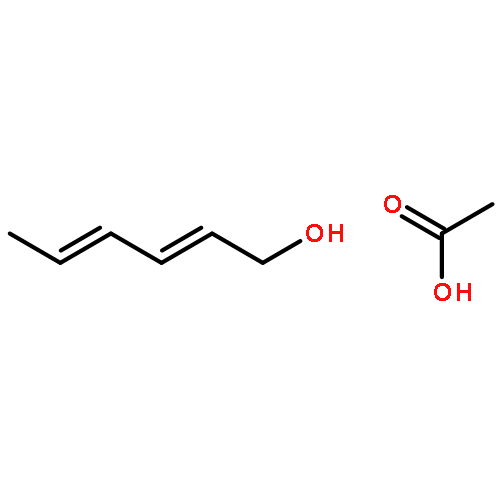
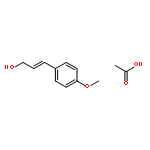
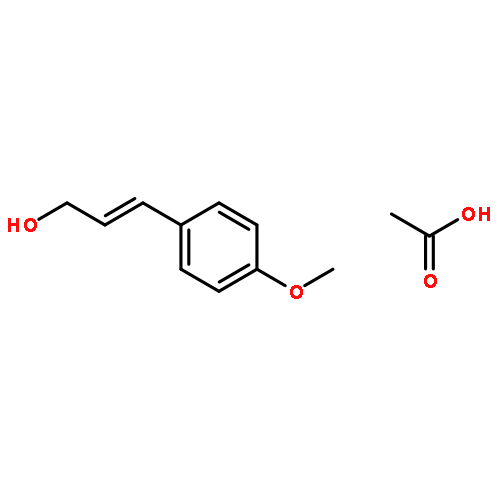




![Benzenamine, N,N-dimethyl-4-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3735000/41652-92-0.png)
![Benzenamine, N,N-dimethyl-4-[(2E)-3-phenyl-2-propen-1-yl]-](/data/chemimg/3735000/41652-92-0_b.png)










![Benzene, [(2E)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1790100/21488-84-6.png)
![Benzene, [(2E)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1790100/21488-84-6_b.png)
![Benzene, [(2Z)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1790400/21488-83-5.png)
![Benzene, [(2Z)-3,7-dimethyl-2,6-octadien-1-yl]-](/data/chemimg/1790400/21488-83-5_b.png)
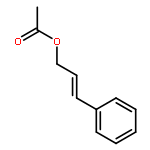
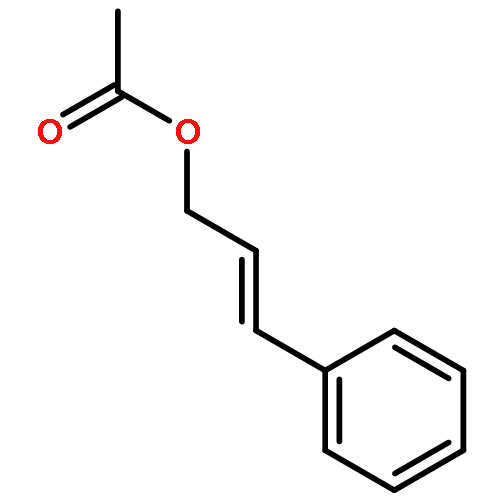

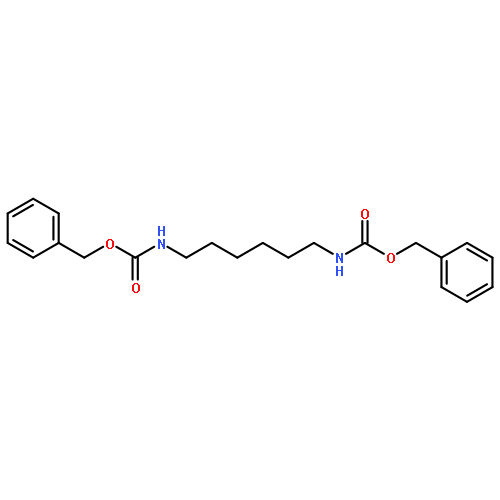




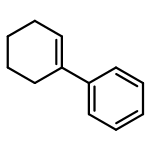
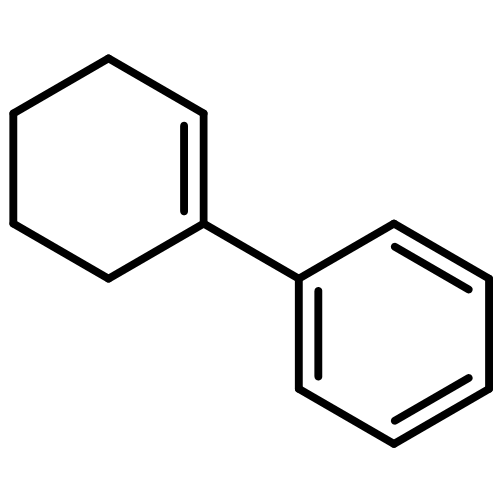


![Benzene, 1,1'-[(1E)-3-methyl-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/7400/7302-01-4.png)
![Benzene, 1,1'-[(1E)-3-methyl-1-propene-1,3-diyl]bis-](http://img.cochemist.com/ccimg/7400/7302-01-4_b.png)
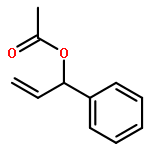
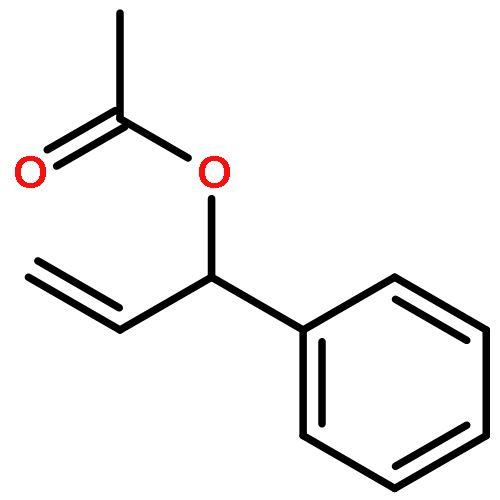











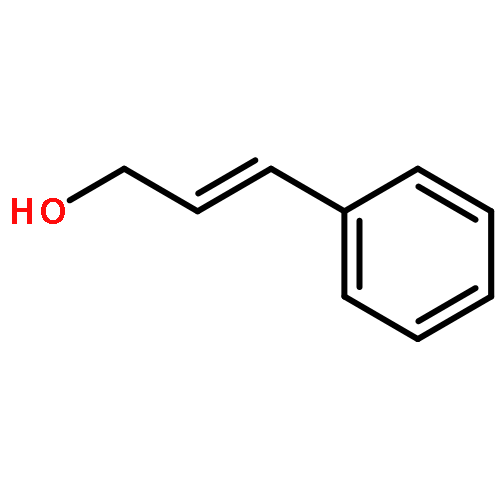




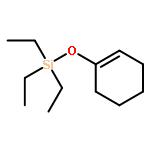

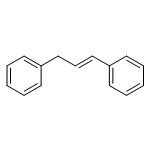
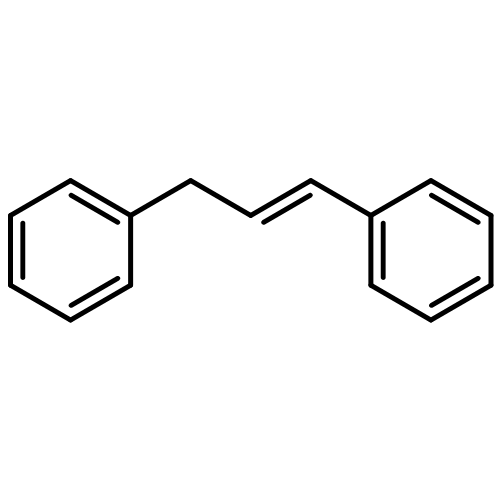


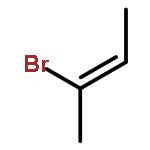
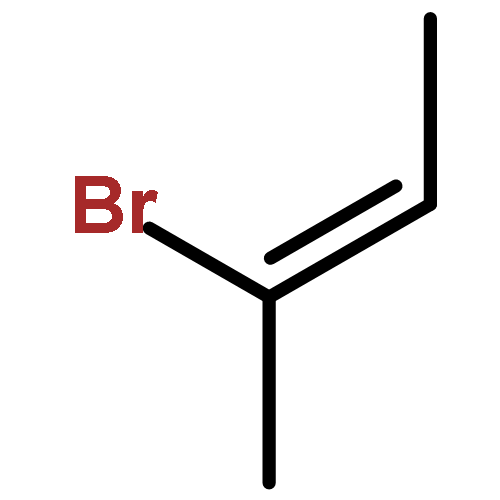





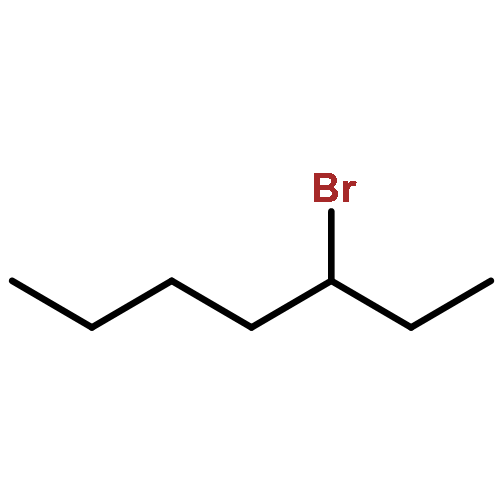


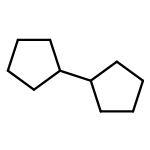
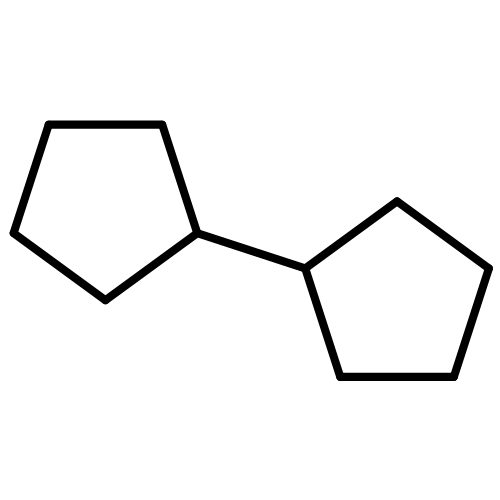


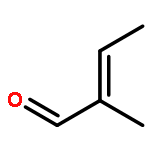
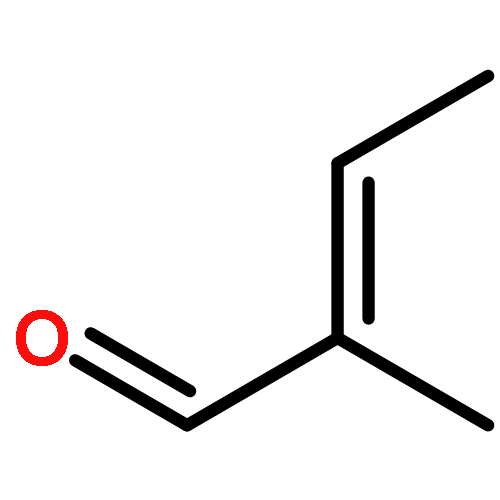


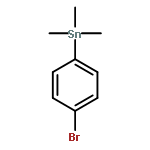
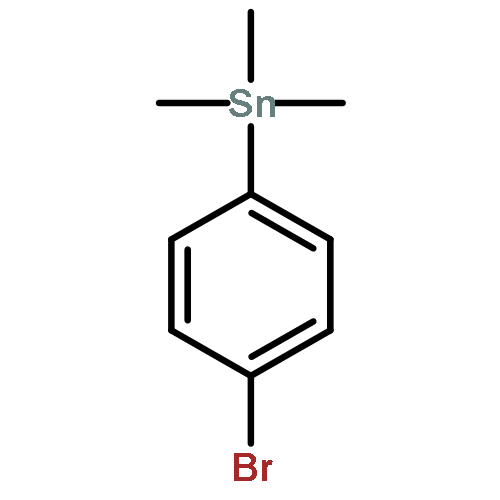
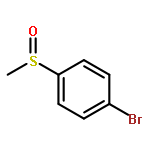
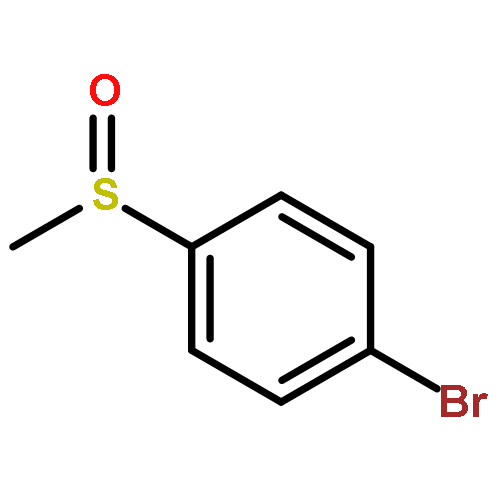

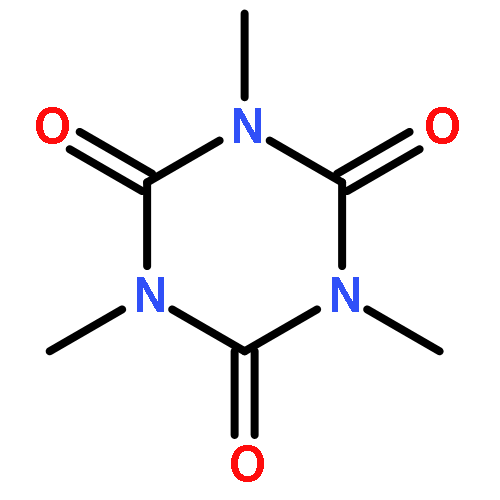


![N-ETHYL-N-[(7-HYDROXY-4-OXO-2-PHENYL-4H-CHROMEN-8-YL)METHYL]ETHAN<WBR />AMINIUM CHLORIDE](http://img.cochemist.com/ccimg/400/351-70-2.png)
![N-ETHYL-N-[(7-HYDROXY-4-OXO-2-PHENYL-4H-CHROMEN-8-YL)METHYL]ETHAN<WBR />AMINIUM CHLORIDE](http://img.cochemist.com/ccimg/400/351-70-2_b.png)