Co-reporter:Xiaomeng Liu, Yayan Tong, Ajing Wang, Xiaopeng Xuan
Journal of Molecular Liquids 2017 Volume 246(Volume 246) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.molliq.2017.09.069
•The crystal structure of 1,3-bis(carboxymethyl)imidazolium nitrate was given.•The OH…O and CH…O hydrogen bonds were found and analyzed.•FT-IR and Raman spectra of 1,3-bis(carboxymethyl)imidazolium nitrate were determined•Thermal stability of 1,3-bis(carboxymethyl)imidazolium nitrate was discussed.In this paper, 1,3-bis(carboxymethyl)imidazolium nitrate was prepared, and its structure was also determined by a single-crystal X-ray diffraction. Its FT-infrared and Raman spectra were measured and reported. The crystal structure and the type of OH…O hydrogen bonds were analyzed on the basis of the vibrational spectroscopy and Density Functional Theory (DFT) calculation. Additionally, thermal stability of 1,3-bis(carboxymethyl)imidazolium nitrate and its decomposition process were discussed.Download high-res image (63KB)Download full-size image
Co-reporter:Rui Liang, Fangfang Yue, Yuting Wang, Yongkang Guo, Xiaopeng Xuan
Journal of Molecular Structure 2016 Volume 1119() pp:301-307
Publication Date(Web):5 September 2016
DOI:10.1016/j.molstruc.2016.04.067
•Three new polymeric complexes based on triangular 1,3-BPEP were synthesized.•Diversity structures were obtained due to the different molecular lengths of carboxylic acid.•Thermal and fluorescence properties of these complexes were investigated.Three new polymeric complexes [Cd3(1,3-BPEB)3(1,4-BDC)3·2H2O]n (1), [Cd2(1,3-BPEB)2(4,4′-BPDC)2]n (2) and [Cd2(1,3-BPEB)2(4,4′-STDC)2]n (3) (1,3-BPEB = 1,3-bis[2-(4′-pyridyl)ethenyl]benzene, 1,4-BDC = 1,4-benzenedicarboxylic acid, 4,4′-BPDC = 4,4′-biphenyldicarboxylic acid and 4,4′-STDC = 4,4′-stilbenedicarboxylic acid) have been prepared by the solvothermal reaction of 1,3-BPEB, Cd(NO3)2·4H2O and dicarboxylic acids. Single-crystal X-ray diffraction reveals that these complexes have novel complicated structures. Complex 1 is a 3-D network structure by linking Cd atoms with 1,4-BDC and 1,3-BPEB ligands. Complex 2 shows a 3-D interpenetrating layered structure formed by three networks which derived from bridged Cd atoms with 4,4′-BPDC and 1,3-BPEB ligands. Complex 3 has a fivefold interpenetrating 3-D coordination framework by linking Cd atoms with 4,4′-STDC and 1,3-BPEB ligands. The dicarboxylic acids afford different coordination modes to bind Cd atoms in these three complexes. Additionally, these compounds were further characterized by PXRD, FT-IR spectroscopy and thermogravimetric (TG) analysis. Their fluorescence spectra were also determined and analyzed in the solid state at room temperature.
Co-reporter:Ajing Wang, Yang Zhao, Xiaomeng Liu, Liangliang Chang, Xiaopeng Xuan
Journal of Fluorine Chemistry 2016 Volume 186() pp:7-11
Publication Date(Web):June 2016
DOI:10.1016/j.jfluchem.2016.04.003
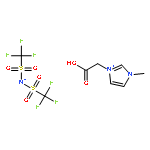
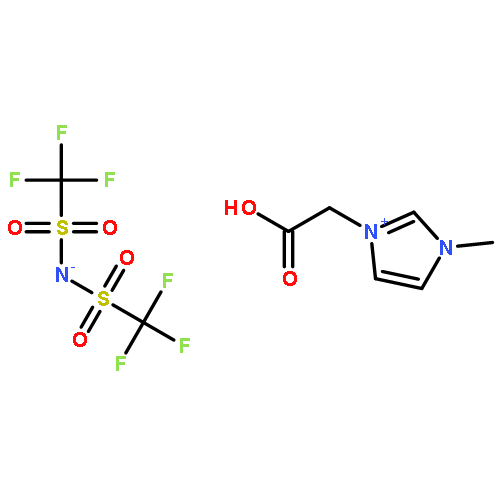
•The crystal structure of 1,3-bis(carboxymethyl)imidazolium bis(trifluoromethylsulfonyl)imide organic salt was determined.•Its solubility in water is higher by about 10 ∼ 20 times than that of N–alkyl-imidazolium TFSI.•High designability of ionic liquid was verified.The bis(trifluoromethylsulfonyl)imide anion (TFSI) is one of the common anions forming hydrophobic ionic liquids at room temperature. In this work, through an introduction of two carboxyl groups into the imidazolium cation, a hydrophilic 1,3-bis(carboxymethyl)imidazolium bis(trifluoromethylsulfonyl)imide (BCITFSI) organic salt was synthesized and its crystal structure, vibrational spectra, physical-chemical properties such as thermal stability, pKa and solubility in water were experimentally determined. Present results are suggesting a possibility to tune the properties of ionic liquids by an appropriate change in cation structure, leading to new “designer solvents”.We synthesized a hydrophilic 1,3-bis(carboxymethyl)imidazolium bis(trifluoromethylsulfonyl)imide organic salt by the introduction of two carboxyl groups into the imidazolium cation, and determined its crystal structure, FT-IR and Raman spectra, and thermal properties. Its solubility in water is higher by about 10 ∼ 20 times than that of the common N–alkyl-imidazolium TFSI ionic liquid. This result indicates that the ionic liquids have high designability, and can be used for a specific purpose by the modification of physical and chemical properties.
Co-reporter:A. J. Wang;H. Zhang;J. L. Gao;X. P. Xuan
Russian Journal of Coordination Chemistry 2016 Volume 42( Issue 4) pp:278-284
Publication Date(Web):2016 April
DOI:10.1134/S1070328416040060
Four novel metal–organic frameworks, [Cu(Tmp)2(H2O)] · NO3 (I) (Tmp = 3,4,7,8-tetramethyl-1,10-phenanthroline), [Mn(Tmp)2(H2O)2] · 2NO3 · H2O (II), [Pb(Tmp)(CH3COO)2] · 3H2O (III) and [Zn(Tmp)2(H2O)2] · 2NO3 · 2H2O (IV), have been synthesized and characterized by single crystal X-ray diffraction (CIF files CCDC nos. 88362–88365 for I–IV, respectively), IR spectroscopy, elemental analysis and thermogravimetric analysis. Both I and II complexes are crystallized in monoclinic system with space groups C2/c, P21/c, respectively, while III and IV complexes are crystallized in triclinic system with space groups \(P\overline 1 \). Generally, these crystal structures are stabilized by O–H···O hydrogen bonds and π–π interactions between the phenanthroline rings of neighboring molecules. Thermogravimetric analyses of compounds I–IV display considerable thermal stability.
Co-reporter:Chunyan Li, Fenghua Cui, Heng Zhang, Xiaopeng Xuan
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015 Volume 134() pp:367-371
Publication Date(Web):5 January 2015
DOI:10.1016/j.saa.2014.06.080
•(C6H11N2)[ZnBr3(C6H6N2O)] was synthesized under ionothermal condition.•Crystal structure, TGA, fluorescence and FT-IR and Raman spectra are reported.•The ionic liquid is not only as a reaction solvent but also a coordinated ligand.The zinc (II) complex with nicotinamide, (C6H11N2)[ZnBr3(C6H6N2O)], was prepared under ionothermal condition by using the ionic liquid 1-ethyl-3-methylimidazolium bromide ([EMIM]Br) as a solvent. At the same time, [EMIM]Br also functions as a structure-directing agent, leading to a framework structure different from those obtained by the conventional methods. Single-crystal X-ray analysis revealed that the coordinated compound crystallizes in monoclinic space group P2(1)/c, and the Zn (II) ion is four-coordinated by one pyridine ring N atom and three bromide anions in a slightly distorted tetrahedron arrangement. The [EMIM]+ cations acting as the extra framework charge balancing species occupy the channels of this asymmetric unit. In the crystal structure, intermolecular NH⋯Br and NH⋯O hydrogen bonds link the molecules to form a supramolecular structure. In addition, this compound was further characterized by FT-IR and Raman spectroscopic techniques, and the observed important bands were assigned. Thermogravimetric analysis (TG), Differential Scanning Calorimetry (DSC) and fluorescent properties of solid samples were also studied at room temperature.Graphical abstract
Co-reporter:Zhi-Hui Zhang;Qi Zhang;Qing-Qing Zhang;Chen Chen;Ming-Yang He;Qun Chen;Guo-Qiang Song;Xiao-Peng Xuan ;Xian-Feng Huang
Acta Crystallographica Section B 2015 Volume 71( Issue 4) pp:437-446
Publication Date(Web):
DOI:10.1107/S2052520615011191
The cocrystallization of lomefloxacin (Lf) with barbituric acid (HBA) and/or isophthalic acid (H2ip) leads to novel binary and ternary salts via hydrogen-bonding recognition. X-ray single-crystal diffraction analyses show that zwitterionic lomefloxacin can adjust itself to fulfill a different supramolecular array in either binary salts or ternary salt co-crystals, formulated as [HLf]·[Hip]·H2O (1), [HLf]·[BA]·[HBA]·H2O (2) and [HLf]·[BA]·[H2ip]·CH3OH·H2O (3). These pharmaceutical agents present uniform charge-assisted hydrogen-bonding networks between HLf cations and acidic coformers with the lattice capturing water molecules. Structural comparison of (2) and (3) indicated that a delicate balance of geometries and hydrogen-bonding partners is required for stacking to favor the formation of ternary salt co-crystals. Cocrystallization was able to overcome the water insolubility of lomefloxacin. Both the salt co-crystals display enhanced solubility and better pharmaceutical applicability.
Co-reporter:Wei Gao, Yong Tian, Xiaopeng Xuan
Journal of Molecular Graphics and Modelling 2015 60() pp: 118-123
Publication Date(Web):July 2015
DOI:10.1016/j.jmgm.2015.04.002
•The nature of non-covalent interaction between cation–cation π–π stacking is explored theoretically.•Such interaction is different to common π–π interaction.•The occurrence of the interactions is attributed to a few contacts of CH and halide.The cation–cation π–π stacking is uncommon but it is essential for the understanding of some supramolecular structures. We explore theoretically the nature of non-covalent interaction occurring in the stacked structure within modeled clusters of 1,3-dimethylimidazolium and halide. The evidences of the energy decomposition analysis (EDA) and reduced density gradient (RDG) approach are different from those of common π–π interaction. Isosurfaces with RDG also illustrate the strength of the titled π–π interaction and their region. Additionally, we find that the occurrence of this interaction is attributed to a few CH···X interactions, as depicted using atom in molecule (AIM) method. This work presents a clear picture of the typical cation–cation π–π interaction and can serve to advance the understanding of this uncommon interaction.Reduced density gradient isosurface map (left) and atom in molecule (right) of the intermolecular interactions in the [Br][DMIM]···[DMIM][Br] dimmers.
Co-reporter:Xiao-Peng Xuan, Liang-Liang Chang, Heng Zhang, Na Wang and Yang Zhao
CrystEngComm 2014 vol. 16(Issue 14) pp:3040-3046
Publication Date(Web):17 Jan 2014
DOI:10.1039/C3CE42492H
The crystal structures of six COOH-functionalized imidazolium ionic liquids (ILs) were determined, and different hydrogen bonds were found in the hydrophobic and hydrophilic ionic liquids. For the hydrophilic COOH-functionalized ILs based on halide anions, the classic O–H⋯Cl− or O–H⋯Br− hydrogen bond was observed by single crystal X-ray diffraction, whereas the carboxyl group dimer was present for the hydrophobic COOH-functionalized ILs based on the (CF3SO2)2N− (TFSI) and PF6− anions. Parallel vibrational spectroscopic studies and DFT calculations have also demonstrated the difference in the hydrogen bond structures. The relationship between the solubility of the ionic liquids in water and their structures was discussed.
Co-reporter:Xiaopeng Xuan, Na Wang, Zaikun Xue
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2012 Volume 96() pp:436-443
Publication Date(Web):October 2012
DOI:10.1016/j.saa.2012.05.051
In this paper, the structure of 1-carboxymethyl-3-methylimidazolium chloride was studied by X-ray diffraction, density functional theory, and FT–IR and Raman spectroscopic techniques for the first time. Title compound crystallizes in the orthorhombic space group Pca21 with the cell dimensions a = 13.445 (6) Å, b = 6.382 (3) Å, c = 9.727 (5) Å and V = 834.6 (7) Å3. All the geometrical parameters have been calculated using by B3LYP with 6–311G++(d,p) basis set. Optimized geometries have been compared with the experimental data, and the hydrogen bond and short contact interactions were discussed. The vibrational frequencies, infrared intensities and Raman scattering activities of the title compound were calculated at the same level. The observed bands were assigned based on the theoretical calculations. The scaled vibrational frequencies seem to coincide with the experimental data with acceptable deviations.Graphical abstractHighlights► Title compound is the simplest imidazolium salt containing one carboxyl group. ► Crystal structure and FT–IR and Raman spectra of title compound are reported. ► Molecular geometric analysis and vibrational assignments were carried out.
Co-reporter:Xiaopeng Xuan, Meng Guo, Yuanchao Pei, Yong Zheng
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2011 Volume 78(Issue 5) pp:1492-1499
Publication Date(Web):May 2011
DOI:10.1016/j.saa.2011.01.039
In order to deepen the understanding of the cation–anion interaction in ionic liquids, the structures of cation, anions, and cation–anion ion-pairs of 1-allyl-3-methylimidazolium-based ionic liquids are optimized using density functional theory (DFT), and their most stable geometries are discussed. The structural parameters, hydrogen bonds and interaction energies of 1-allyl-3-methylimidazolium dicyanamide ([Amim]DCA), 1-allyl-3-methylimidazolium chloride ([Amim]Cl), 1-allyl-3-methylimidazolium formate ([Amim]FmO) and 1-allyl-3-methylimidazolium acetate ([Amim]AcO) ion pairs are studied. The vibrational frequencies of [Amim]DCA and [Amim]Cl have been calculated and scaled values have been compared with experimental FT-IR and FT-Raman spectra. The complete assignments were performed on the basis of the potential energy distribution (PED) of the vibrational modes.Graphical abstractResearch highlights► 1-allyl-3-methylimidazolium–based ionic liquids ► The hydrogen bonding interactions between the anions and the H atoms of CC–H group in ring and of –CH2– group in alkyl side chain. ► The rotation of allyl group can lead the low viscosity of Amim+–based ion pairs. ► The vibrational spectra of 1-allyl-3-methylimidazolium chloride and 1-allyl-3-methylimidazolium dicyanamide were reported.
Co-reporter:Xiaopeng Xuan, Xinsheng Wang, Na Wang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2011 Volume 79(Issue 5) pp:1091-1098
Publication Date(Web):September 2011
DOI:10.1016/j.saa.2011.04.024
Molecular structure and vibrational spectra of 1,4-dihydroxyanthraquinone (1,4-DAQ) are studied theoretically and experimentally. FT-infrared and Raman spectra of 1,4-DAQ are recorded in solid phase in regions of 4000–400 and 3500–100 cm−1, respectively. The geometry of 1,4-DAQ is optimized by B3LYP and B3PW91 methods, and the most stable structure with C2v point group is found. The harmonic vibration frequencies, infrared intensities, and the Raman scattering activities of the compound are calculated, analyzed, and compared with experimental data. Our calculated frequencies agree well with the experimental results.Graphical abstractHighlights► FT-IR and Raman spectra are reported for 1,4-dihydroxyanthraquinone. ► 1,4-Dihydroxyanthraquinone in gas phase has a stable structure with C2V symmetry. ► The spectroscopic data of 1,4-dihydroxyanthraquinone are assigned and compared with the results calculated at B3LYP/6-311++G(d,p) level.
Co-reporter:Xiaopeng Xuan, Yingling Wang, Na Wang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2011 Volume 81(Issue 1) pp:236-241
Publication Date(Web):15 October 2011
DOI:10.1016/j.saa.2011.05.110
The infrared (4000–400 cm−1) and Raman spectra (3700–100 cm−1) of liquid S-phenyl thioacetate have been recorded. Molecular geometry, vibrational frequencies and the corresponding assignments were performed by density funtional theory (DFT) using the 6-311++G(d,p) basis set. Two conformers have been identified. One is syn form where the carbonyl group is on the same side of the phenyl ring and the other is the anti form. The energy difference between these two configurations is about 1.63 kcal mol−1 at B3LYP/6-311++G(d,p) level. By utilizing the more stable syn conformer, a complete assignment of the observed frequencies is given according to the total energy distribution of the vibrational modes. The general agreements between the observed and the calculated frequencies are shown.Graphical abstractHighlights► FT-IR and Raman spectra are reported for S-phenyl thioacetate. ► The syn conformer of S-phenyl thioacetate with C1 symmetry is most stable. ► The IR and Raman spectra of S-phenyl thioacetate are recorded and assigned.


















![1H,3H-Naphtho[1,8-cd]pyran-1,3-dione,5-amino-](http://img.cochemist.com/ccimg/23300/23204-38-8.png)
![1H,3H-Naphtho[1,8-cd]pyran-1,3-dione,5-amino-](http://img.cochemist.com/ccimg/23300/23204-38-8_b.png)

