Co-reporter:Christopher R. Coxon, Elizabeth Anscombe, Suzannah J. HarnorMathew P. Martin, Benoit Carbain, Bernard T. Golding, Ian R. Hardcastle, Lisa K. Harlow, Svitlana Korolchuk, Christopher J. Matheson, David R. Newell, Martin E. M. Noble, Mangaleswaran Sivaprakasam, Susan J. Tudhope, David M. Turner, Lan Z. Wang, Stephen R. Wedge, Christopher Wong, Roger J. Griffin, Jane A. Endicott, Céline Cano
Journal of Medicinal Chemistry 2017 Volume 60(Issue 5) pp:
Publication Date(Web):December 22, 2016
DOI:10.1021/acs.jmedchem.6b01254
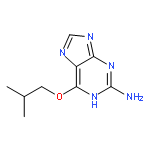
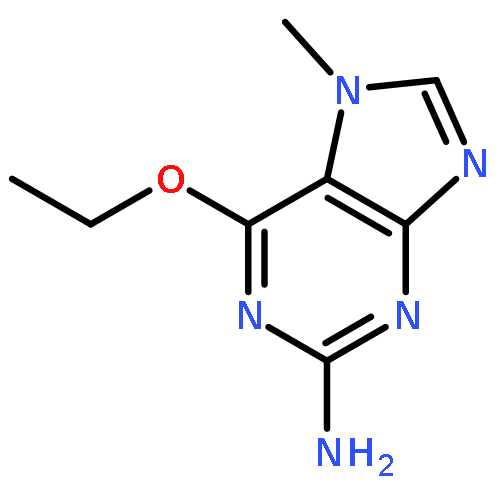
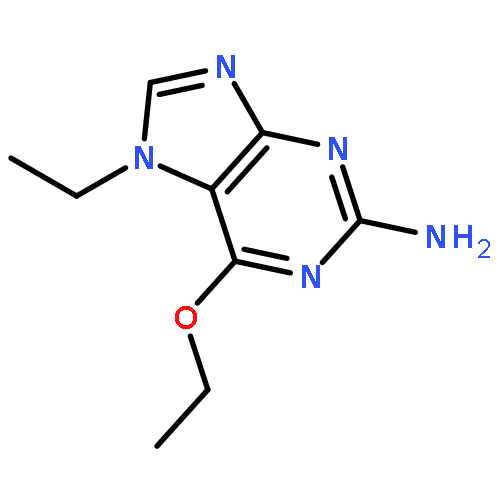
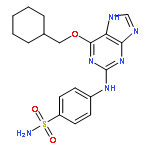
Purines and related heterocycles substituted at C-2 with 4′-sulfamoylanilino and at C-6 with a variety of groups have been synthesized with the aim of achieving selectivity of binding to CDK2 over CDK1. 6-Substituents that favor competitive inhibition at the ATP binding site of CDK2 were identified and typically exhibited 10–80-fold greater inhibition of CDK2 compared to CDK1. Most impressive was 4-((6-([1,1′-biphenyl]-3-yl)-9H-purin-2-yl)amino) benzenesulfonamide (73) that exhibited high potency toward CDK2 (IC50 0.044 μM) but was ∼2000-fold less active toward CDK1 (IC50 86 μM). This compound is therefore a useful tool for studies of cell cycle regulation. Crystal structures of inhibitor–kinase complexes showed that the inhibitor stabilizes a glycine-rich loop conformation that shapes the ATP ribose binding pocket and that is preferred in CDK2 but has not been observed in CDK1. This aspect of the active site may be exploited for the design of inhibitors that distinguish between CDK1 and CDK2.
Co-reporter:Céline Cano ; Kappusamy Saravanan ; Chris Bailey ; Julia Bardos ; Nicola J. Curtin ; Mark Frigerio ; Bernard T. Golding ; Ian R. Hardcastle ; Marc G. Hummersone ; Keith A. Menear ; David R. Newell ; Caroline J. Richardson ; K. Shea ; Graeme C. M. Smith ; Pia Thommes ; Attilla Ting ;Roger J. Griffin
Journal of Medicinal Chemistry 2013 Volume 56(Issue 16) pp:6386-6401
Publication Date(Web):July 15, 2013
DOI:10.1021/jm400915j
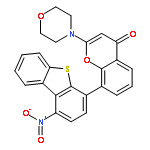
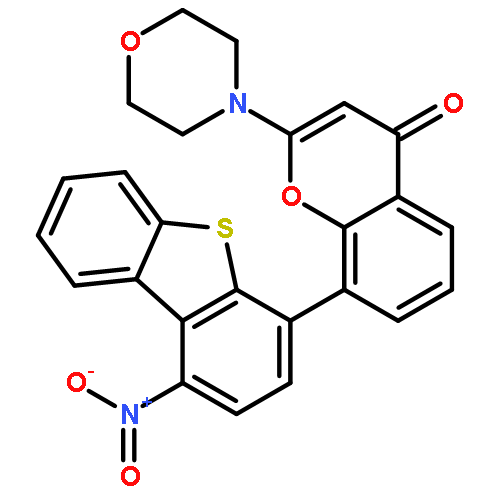
Analogues of (dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-one (NU7441), a potent inhibitor of DNA-dependent protein kinase (DNA-PK; IC50 = 42 ± 2 nM), have been synthesized in which water-solubilizing groups [NHCO(CH2)nNR1R2, where n = 1 or 2 and the moiety R1R2N was derived from a library of primary and secondary amines, e.g., morpholine] were placed at the 1-position. Several of the newly synthesized compounds exhibited high potency against DNA-PK and potentiated the cytotoxicity of ionizing radiation (IR) in vitro 10-fold or more (e.g., 2-(4-ethylpiperazin-1-yl)-N-(4-(2-morpholino-4-oxo-4H-chromen-8-yl)dibenzo[b,d]thio-phen-1-yl)acetamide, 39; DNA-PK IC50 = 5.0 ± 1 nM, IR dose modification ratio = 13). Furthermore, 39 was shown to potentiate not only IR in vitro but also DNA-inducing cytotoxic anticancer agents, both in vitro and in vivo. Counter-screening against other members of the phosphatidylinositol 3-kinase (PI-3K) related kinase (PIKK) family unexpectedly revealed that some of the compounds were potent mixed DNA-PK and PI-3K inhibitors.
Co-reporter:Kate M. Clapham, Tommy Rennison, Gavin Jones, Faye Craven, Julia Bardos, Bernard T. Golding, Roger J. Griffin, Karen Haggerty, Ian R. Hardcastle, Pia Thommes, Attilla Ting and Céline Cano
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 33) pp:6747-6757
Publication Date(Web):16 Jul 2012
DOI:10.1039/C2OB26035B
Substitution at the 7-position of the chromen-4-one pharmacophore of 8-(dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-one NU7441, a potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor, with allyl, n-propyl or methyl enabled the resolution by chiral HPLC of atropisomers. Biological evaluation against DNA-PK of each pair of atropisomers showed a marked difference in potency, with biological activity residing exclusively in the laevorotatory enantiomer.
Co-reporter:Kate M. Clapham, Julia Bardos, M. Raymond V. Finlay, Bernard T. Golding, Edward J. Griffen, Roger J. Griffin, Ian R. Hardcastle, Keith A. Menear, Attilla Ting, Paul Turner, Gail L. Young, Céline Cano
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 3) pp:966-970
Publication Date(Web):1 February 2011
DOI:10.1016/j.bmcl.2010.12.047
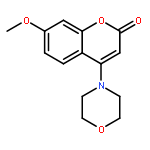
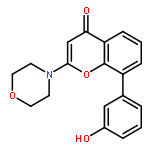
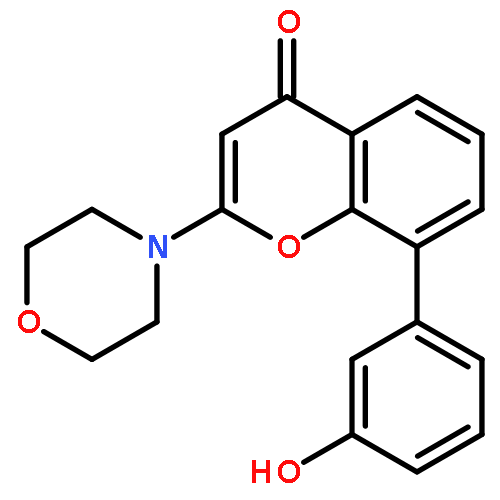
Introduction of an O-alkoxyphenyl substituent at the 8-position of the 2-morpholino-4H-chromen-4-one pharmacophore enabled regions of the ATP-binding site of DNA-dependent protein kinase (DNA-PK) to be probed further. Structure–activity relationships have been elucidated for inhibition of DNA-PK and PI3K (p110α), with N-(2-(cyclopropylmethoxy)-4-(2-morpholino-4-oxo-4H-chromen-8-yl)phenyl)-2-morpholinoacetamide 11a being identified as a potent and selective DNA-PK inhibitor (IC50 = 8 nM).Introduction of an O-alkoxyphenyl substituent at the 8-position of the 2-morpholino-4H-chromen-4-one pharmacophore enabled regions of the ATP-binding site of DNA-dependent protein kinase (DNA-PK) to be probed further.
Co-reporter:Céline Cano ; Olivier R. Barbeau ; Christine Bailey ; Xiao-Ling Cockcroft ; Nicola J. Curtin ; Heather Duggan ; Mark Frigerio ; Bernard T. Golding ; Ian R. Hardcastle ; Marc G. Hummersone ; Charlotte Knights ; Keith A. Menear ; David R. Newell ; Caroline J. Richardson ; Graeme C. M. Smith ; Ben Spittle ;Roger J. Griffin
Journal of Medicinal Chemistry 2010 Volume 53(Issue 24) pp:8498-8507
Publication Date(Web):November 16, 2010
DOI:10.1021/jm100608j
Following the discovery of dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-one (NU7441) (Leahy, J. J. J.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Richardson, C.; Rigoreau, L.; Smith, G. C. M. Bioorg. Med. Chem. Lett. 2004, 14, 6083−6087) as a potent inhibitor (IC50 = 30 nM) of DNA-dependent protein kinase (DNA-PK), we have investigated analogues in which the chromen-4-one core template has been replaced by aza-heterocyclic systems: 9-substituted 2-morpholin-4-ylpyrido[1,2-a]pyrimidin-4-ones and 8-substituted 2-morpholin-4-yl-1H-quinolin-4-ones. The 8- and 9-substituents were either dibenzothiophen-4-yl or dibenzofuran-4-yl, which were each further substituted at the 1-position with water-solubilizing groups [NHCO(CH2)nNR1R2, where n = 1 or 2 and the moiety R1R2N was derived from a library of primary and secondary amines (e.g., morpholine)]. The inhibitors were synthesized by employing a multiple-parallel approach in which the two heterocyclic components were assembled by Suzuki−Miyaura cross-coupling. Potent DNA-PK inhibitory activity was generally observed across the compound series, with structure−activity studies indicating that optimal potency resided in pyridopyrimidin-4-ones bearing a substituted dibenzothiophen-4-yl group. Several of the newly synthesized compounds (e.g., 2-morpholin-4-yl-N-[4-(2-morpholin-4-yl-4-oxo-4H-pyrido[1,2-a]pyrimidin-9-yl)dibenzothiophen-1-yl]acetamide) combined high potency against the target enzyme (DNA-PK IC50 = 8 nM) with promising activity as potentiators of ionizing radiation-induced cytotoxicity in vitro.
Co-reporter:Céline Cano, Bernard T. Golding, Karen Haggerty, Ian R. Hardcastle, Marcus Peacock and Roger J. Griffin
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 8) pp:1922-1928
Publication Date(Web):01 Mar 2010
DOI:10.1039/B926245H
Substitution at the 3-position of the dibenzothiophen-4-yl ring of 8-(dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-one NU7441, a potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor, with propyl, allyl or methyl enabled the separation by chiral HPLC of atropisomers. This is a consequence of restricted rotation about the dibenzothiophene–chromenone bond. Biological evaluation against DNA-PK of the pairs of atropisomers showed a marked difference in potency, with only one enantiomer being biologically active.
Co-reporter:Kate M. Clapham, Tommy Rennison, Gavin Jones, Faye Craven, Julia Bardos, Bernard T. Golding, Roger J. Griffin, Karen Haggerty, Ian R. Hardcastle, Pia Thommes, Attilla Ting and Céline Cano
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 33) pp:NaN6757-6757
Publication Date(Web):2012/07/16
DOI:10.1039/C2OB26035B
Substitution at the 7-position of the chromen-4-one pharmacophore of 8-(dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-one NU7441, a potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor, with allyl, n-propyl or methyl enabled the resolution by chiral HPLC of atropisomers. Biological evaluation against DNA-PK of each pair of atropisomers showed a marked difference in potency, with biological activity residing exclusively in the laevorotatory enantiomer.
Co-reporter:Céline Cano, Bernard T. Golding, Karen Haggerty, Ian R. Hardcastle, Marcus Peacock and Roger J. Griffin
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 8) pp:NaN1928-1928
Publication Date(Web):2010/03/01
DOI:10.1039/B926245H
Substitution at the 3-position of the dibenzothiophen-4-yl ring of 8-(dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-one NU7441, a potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor, with propyl, allyl or methyl enabled the separation by chiral HPLC of atropisomers. This is a consequence of restricted rotation about the dibenzothiophene–chromenone bond. Biological evaluation against DNA-PK of the pairs of atropisomers showed a marked difference in potency, with only one enantiomer being biologically active.








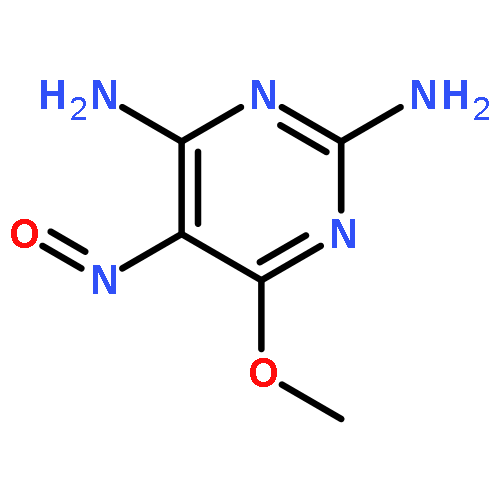
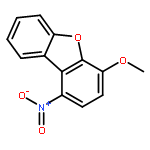
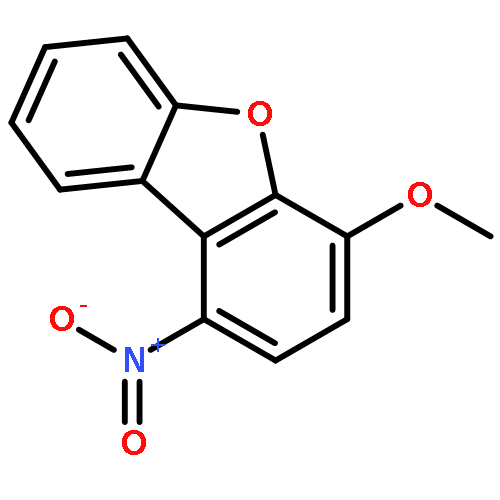




![Glycine, N-[[[3-cyano-4-(4-fluorophenyl)-6-phenyl-2-pyridinyl]thio]acetyl]-](http://img.cochemist.com/ccimg/353500/353463-50-0.png)
![Glycine, N-[[[3-cyano-4-(4-fluorophenyl)-6-phenyl-2-pyridinyl]thio]acetyl]-](http://img.cochemist.com/ccimg/353500/353463-50-0_b.png)
![Glycine, N-[[(3-cyano-4,6-dimethyl-2-pyridinyl)thio]acetyl]-](http://img.cochemist.com/ccimg/385500/385417-58-3.png)
![Glycine, N-[[(3-cyano-4,6-dimethyl-2-pyridinyl)thio]acetyl]-](http://img.cochemist.com/ccimg/385500/385417-58-3_b.png)







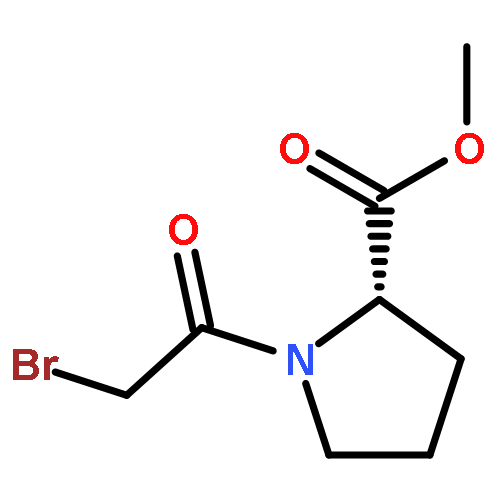













![dibenzo[b,d]furan-4-ol](http://img.cochemist.com/ccimg/19300/19261-06-4.png)
![dibenzo[b,d]furan-4-ol](http://img.cochemist.com/ccimg/19300/19261-06-4_b.png)
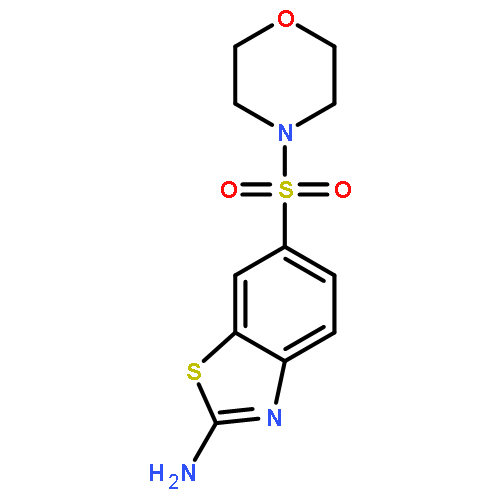
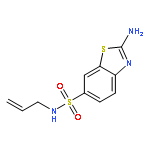
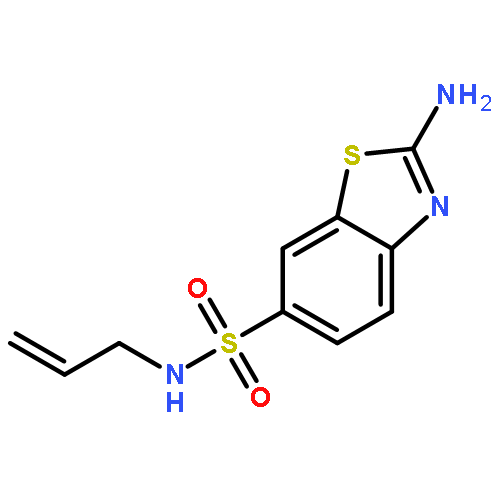
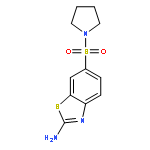
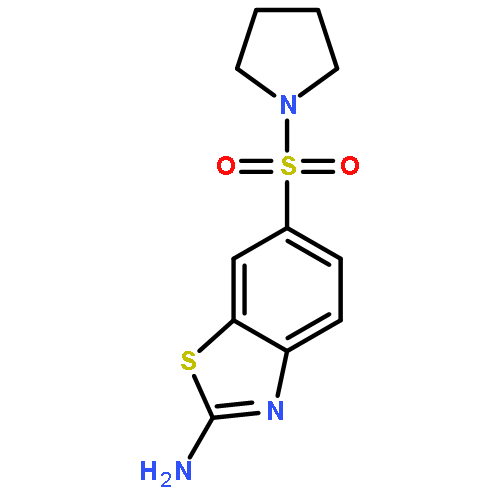
![2-Amino-N,N-diethylbenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18000/17901-14-3.png)
![2-Amino-N,N-diethylbenzo[d]thiazole-6-sulfonamide](http://img.cochemist.com/ccimg/18000/17901-14-3_b.png)






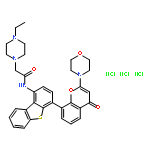
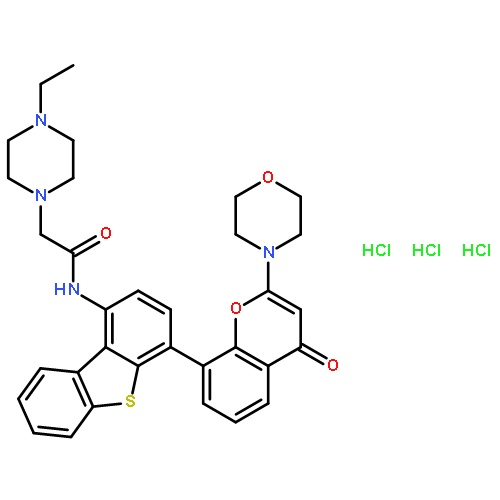
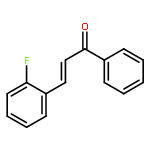


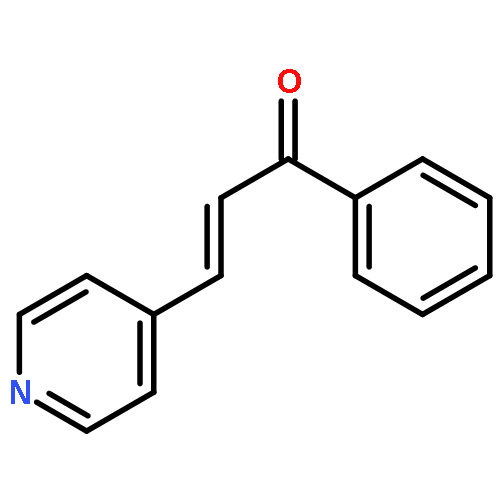


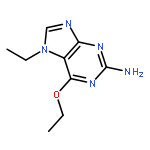











![9H-Purin-2-amine, 6-chloro-9-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/203300/203201-61-0.png)
![9H-Purin-2-amine, 6-chloro-9-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/203300/203201-61-0_b.png)












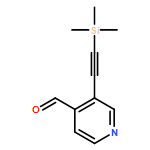
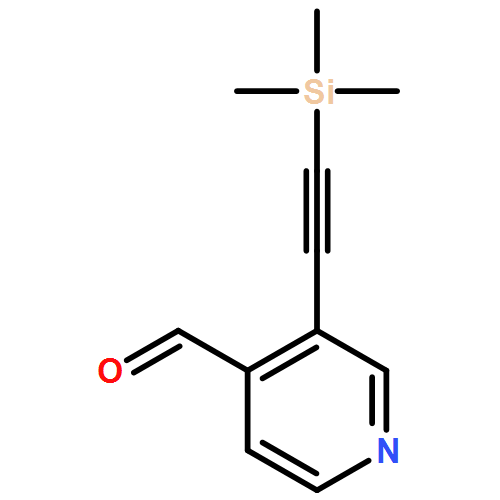

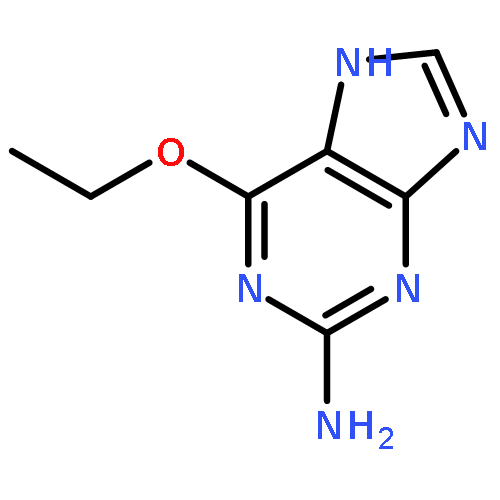


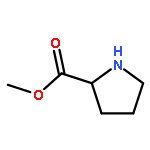












![4H-1-Benzopyran-4-one,2-(4-morpholinyl)-8-[3-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/503500/503468-65-3.png)
![4H-1-Benzopyran-4-one,2-(4-morpholinyl)-8-[3-(phenylmethoxy)phenyl]-](http://img.cochemist.com/ccimg/503500/503468-65-3_b.png)



















![Benzoic acid, 2-[2-(4-morpholinyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/61500/61418-24-4.png)
![Benzoic acid, 2-[2-(4-morpholinyl)-2-oxoethyl]-](http://img.cochemist.com/ccimg/61500/61418-24-4_b.png)




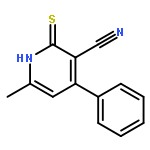





![4-[2-(4-METHYL-1-PIPERAZINYL)ETHOXY]ANILINE](http://img.cochemist.com/ccimg/39000/38948-28-6.png)
![4-[2-(4-METHYL-1-PIPERAZINYL)ETHOXY]ANILINE](http://img.cochemist.com/ccimg/39000/38948-28-6_b.png)


![1H-IMIDAZO[4,5-C]PYRIDINE-5-AMINE, 7-CHLORO-, MONOHYDRATE](http://img.cochemist.com/ccimg/37500/37436-96-7.png)
![1H-IMIDAZO[4,5-C]PYRIDINE-5-AMINE, 7-CHLORO-, MONOHYDRATE](http://img.cochemist.com/ccimg/37500/37436-96-7_b.png)








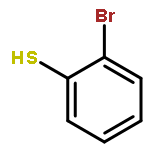
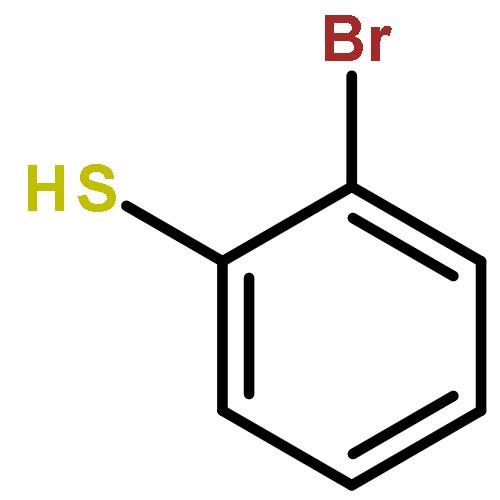




![5,7-Dichloroimidazo[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/57500/57473-32-2.png)
![5,7-Dichloroimidazo[1,2-a]pyrimidine](http://img.cochemist.com/ccimg/57500/57473-32-2_b.png)
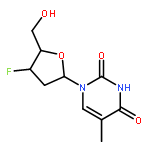
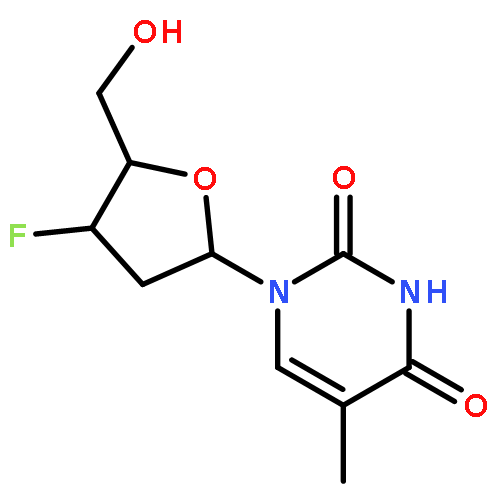
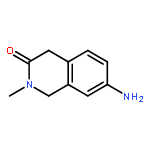



























































![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/144100/144017-74-3.png)
![2-Propen-1-one, 1-phenyl-3-[4-(trifluoromethyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/144100/144017-74-3_b.png)



