Co-reporter:Gaël Tran, Danielle Confair, Kevin D. Hesp, Vincent Mascitti, and Jonathan A. Ellman
The Journal of Organic Chemistry September 1, 2017 Volume 82(Issue 17) pp:9243-9243
Publication Date(Web):August 22, 2017
DOI:10.1021/acs.joc.7b01723
Herein, we report a Rh(I)/bisphosphine/K3PO4 catalytic system allowing for the first time the selective branched C–H alkylation of benzimidazoles with Michael acceptors. Branched alkylation with N,N-dimethyl acrylamide was successfully applied to the alkylation of a broad range of benzimidazoles incorporating a variety of N-substituents and with both electron-rich and -poor functionality displayed at different sites of the arene. Moreover, the introduction of a quaternary carbon was achieved by alkylation with ethyl methacrylate. The method was also shown to be applicable to the C2-selective branched alkylation of azabenzimidazoles.
Co-reporter:Joshua R. Hummel, Jeffrey A. Boerth, and Jonathan A. Ellman
Chemical Reviews July 12, 2017 Volume 117(Issue 13) pp:9163-9163
Publication Date(Web):December 12, 2016
DOI:10.1021/acs.chemrev.6b00661
The transition-metal-catalyzed addition of C–H bonds to carbonyls, imines, and related polarized π bonds has emerged as a particularly efficient and powerful approach for the construction of an incredibly diverse array of heteroatom-substituted products. Readily available and stable inputs are typically employed, and reactions often proceed with very high functional group compatibility and without the production of waste byproducts. Additionally, many transition-metal-catalyzed C–H bond additions to polarized π bonds occur within cascade reaction sequences to provide rapid access to a diverse array of different heterocyclic as well as carbocyclic products. This review highlights the diversity of transformations that have been achieved, catalysts that have been used, and types of products that have been prepared through the transition-metal-catalyzed addition of C–H bonds to carbonyls, imines, and related polarized π bonds.
Co-reporter:Tyler J. Potter, David N. Kamber, Brandon Q. Mercado, and Jonathan A. Ellman
ACS Catalysis January 6, 2017 Volume 7(Issue 1) pp:150-150
Publication Date(Web):December 2, 2016
DOI:10.1021/acscatal.6b03217
The transition-metal-catalyzed C–H bond addition to nitroalkenes has been developed. Very broad nitroalkene scope was observed for this Rh(III)-catalyzed method, including for aliphatic, aromatic, and β,β-disubstituted derivatives. Additionally, various directing groups and both aromatic and alkenyl C–H bonds were effective in this transformation. Representative nitroalkane products were converted to dihydroisoquinolones and dihydropyridones in a single step and in high yield by iron-mediated reduction and in situ cyclization. Moreover, preliminary success in enantioselective Rh(III)-catalyzed C–H bond addition to nitroalkenes was achieved as was X-ray structural characterization of a nitronate intermediate.Keywords: C−H functionalization; dihydroisoquinolone; enantioselective catalysis; nitroalkene; Rh(III) catalysis;
Co-reporter:Caroline Chandra Tjin, Kate D. Otley, Tyler D. Baguley, Pradeep Kurup, Jian Xu, Angus C. Nairn, Paul J. Lombroso, and Jonathan A. Ellman
ACS Central Science December 27, 2017 Volume 3(Issue 12) pp:1322-1322
Publication Date(Web):December 6, 2017
DOI:10.1021/acscentsci.7b00486
Dysregulation of protein tyrosine phosphorylation has been implicated in a number of human diseases, including cancer, diabetes, and neurodegenerative diseases. As a result of their essential role in regulating protein tyrosine phosphorylation levels, protein tyrosine phosphatases (PTPs) have emerged as important yet challenging therapeutic targets. Here we report on the development and application of a glutathione-responsive motif to facilitate the efficient intracellular delivery of a novel class of selenosulfide phosphatase inhibitors for the selective active site directed inhibition of the targeted PTP by selenosulfide exchange with the active site cysteine. The strategy leverages the large difference in extracellular and intracellular glutathione levels to deliver selenosulfide phosphatase inhibitors to cells. As an initial exploration of the prodrug platform and the corresponding selenosulfide covalent inhibitor class, potent and selective inhibitors were developed for two therapeutically relevant PTP targets: the Mycobacterium tuberculosis virulence factor mPTPA and the CNS-specific tyrosine phosphatase, striatal-enriched protein tyrosine phosphatase (STEP). The lead selenosulfide inhibitors enable potent and selective inhibition of their respective targets over a panel of human PTPs and a representative cysteine protease. Kinetic parameters of the inhibitors were characterized, including reversibility of inhibition and rapid rate of GSH exchange at intracellular GSH concentrations. Additionally, active site covalent inhibitor-labeling with an mPTPA inhibitor was rigorously confirmed by mass spectrometry, and cellular activity was demonstrated with a STEP prodrug inhibitor in cortical neurons.
Co-reporter:Yajing Lian, Joshua R. Hummel, Robert G. Bergman, and Jonathan A. Ellman
Journal of the American Chemical Society August 28, 2013 Volume 135(Issue 34) pp:12548-12551
Publication Date(Web):August 19, 2013
DOI:10.1021/ja406131a
We report formal [3 + 3] annulations of aromatic azides with aromatic imines and azobenzenes to give acridines and phenazines, respectively. These transformations proceed through a cascade process of Rh(III)-catalyzed amination followed by intramolecular electrophilic aromatic substitution and aromatization. Acridines can be directly prepared from aromatic aldehydes by in situ imine formation using catalytic benzylamine.
Co-reporter:Dr. Gaël Tran;Dr. Kevin D. Hesp;Dr. Vincent Mascitti; Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2017 Volume 56(Issue 21) pp:5899-5903
Publication Date(Web):2017/05/15
DOI:10.1002/anie.201702409
AbstractA [RhI]/bisphosphine/base catalytic system for the ortho-selective C−H alkylation of azines by acrylates and acrylamides is reported. This catalytic system features an unprecedented complete linear or branched selectivity that is solely dependent on the catalytic base that is used. Complete branched selectivity is even achieved for ethyl methacrylate, which enables the introduction of a quaternary carbon center. Excellent functional group compatibility is demonstrated for both linear and branched alkylations. The operational simplicity and broad scope of this transformation allow for rapid access to functionalized azines of direct pharmaceutical and agrochemical relevance.
Co-reporter:Dr. Kim Søholm Halskov;Dr. Howard S. Roth; Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2017 Volume 56(Issue 31) pp:9183-9187
Publication Date(Web):2017/07/24
DOI:10.1002/anie.201703967
AbstractThe first syntheses of privileged [5,6]-bicyclic heterocycles, with ring-junction nitrogen atoms, by transition metal catalyzed C−H functionalization of C-alkenyl azoles is disclosed. Several reactions are applied to alkenyl imidazoles, pyrazoles, and triazoles to provide products with nitrogen incorporated at different sites. Alkyne and diazoketone coupling partners give azolopyridines with various substitution patterns. In addition, 1,4,2-dioxazolone coupling partners yield azolopyrimidines. Furthermore, the mechanisms for the reactions are discussed and the utility of the developed approach is demonstrated by iterative application of C−H functionalization for the rapid synthesis of a patented drug candidate.
Co-reporter:Dr. Gaël Tran;Dr. Kevin D. Hesp;Dr. Vincent Mascitti; Dr. Jonathan A. Ellman
Angewandte Chemie 2017 Volume 129(Issue 21) pp:5993-5997
Publication Date(Web):2017/05/15
DOI:10.1002/ange.201702409
AbstractA [RhI]/bisphosphine/base catalytic system for the ortho-selective C−H alkylation of azines by acrylates and acrylamides is reported. This catalytic system features an unprecedented complete linear or branched selectivity that is solely dependent on the catalytic base that is used. Complete branched selectivity is even achieved for ethyl methacrylate, which enables the introduction of a quaternary carbon center. Excellent functional group compatibility is demonstrated for both linear and branched alkylations. The operational simplicity and broad scope of this transformation allow for rapid access to functionalized azines of direct pharmaceutical and agrochemical relevance.
Co-reporter:Jeffrey A. Boerth; Dr. Jonathan A. Ellman
Angewandte Chemie 2017 Volume 129(Issue 33) pp:10108-10112
Publication Date(Web):2017/08/07
DOI:10.1002/ange.201705817
AbstractA CoIII-catalyzed three-component coupling of C(sp2)−H bonds, alkynes, and halogenating agents to give alkenyl halides is reported. This transformation proceeds with high regio- and diastereoselectivity, and is effective for a broad range of aryl and alkyl terminal alkynes. Diverse C−H bond partners also exhibit good reactivity for a range of heteroaryl and aryl systems as well as synthetically useful secondary and tertiary amide, urea, and pyrazole directing groups. This multicomponent transformation is also compatible with allenes in place of alkynes to furnish tetrasubstituted alkenyl halides, showcasing the first halo-arylation of allenes.
Co-reporter:Dr. Kim Søholm Halskov;Dr. Howard S. Roth; Dr. Jonathan A. Ellman
Angewandte Chemie 2017 Volume 129(Issue 31) pp:9311-9315
Publication Date(Web):2017/07/24
DOI:10.1002/ange.201703967
AbstractThe first syntheses of privileged [5,6]-bicyclic heterocycles, with ring-junction nitrogen atoms, by transition metal catalyzed C−H functionalization of C-alkenyl azoles is disclosed. Several reactions are applied to alkenyl imidazoles, pyrazoles, and triazoles to provide products with nitrogen incorporated at different sites. Alkyne and diazoketone coupling partners give azolopyridines with various substitution patterns. In addition, 1,4,2-dioxazolone coupling partners yield azolopyrimidines. Furthermore, the mechanisms for the reactions are discussed and the utility of the developed approach is demonstrated by iterative application of C−H functionalization for the rapid synthesis of a patented drug candidate.
Co-reporter:Jeffrey A. Boerth; Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2017 Volume 56(Issue 33) pp:9976-9980
Publication Date(Web):2017/08/07
DOI:10.1002/anie.201705817
AbstractA CoIII-catalyzed three-component coupling of C(sp2)−H bonds, alkynes, and halogenating agents to give alkenyl halides is reported. This transformation proceeds with high regio- and diastereoselectivity, and is effective for a broad range of aryl and alkyl terminal alkynes. Diverse C−H bond partners also exhibit good reactivity for a range of heteroaryl and aryl systems as well as synthetically useful secondary and tertiary amide, urea, and pyrazole directing groups. This multicomponent transformation is also compatible with allenes in place of alkynes to furnish tetrasubstituted alkenyl halides, showcasing the first halo-arylation of allenes.
Co-reporter:Shuming Chen, Vlad Bacauanu, Tobias Knecht, Brandon Q. Mercado, Robert G. Bergman, and Jonathan A. Ellman
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:12664-12670
Publication Date(Web):September 19, 2016
DOI:10.1021/jacs.6b08355
Multisubstituted tropanes and indolizidines have been prepared with high regio- and stereoselectivity by the [3+2] cycloaddition of unstabilized azomethine ylides generated from readily prepared trimethylsilyl-substituted 1,2-dihydropyridines via protonation or alkylation followed by desilylation. Starting from 1,2-dihydropyridines bearing a ring trimethylsilyl substituent at the 6-position, an intermolecular alkylation/desilylation provides endocyclic unstabilized ylides that successfully undergo cycloaddition with a range of symmetrical and unsymmetrical alkyne and alkene dipolarophiles to afford densely substituted tropanes incorporating quaternary carbons in good yields and with high regio- and stereoselectivity. Additionally, an intramolecular alkylation/desilylation/cycloaddition sequence provides convenient and rapid entry to bridged tricyclic tropane skeletons, allowing for five contiguous carbon stereocenters to be set in a single experimental operation and under mild conditions. Starting from 1,2-dihydropyridines with trimethylsilylmethyl groups on nitrogen, protonation followed by desilylation generates exocyclic unstabilized ylides that undergo cycloaddition with unsymmetrical alkynes to give indolizidines with good regio- and stereoselectivity. N-Trimethylsilylmethyl-1,2-dihydropyridines can also be alkylated and subsequently desilylated to give endocyclic unstabilized ylides that undergo intermolecular cycloadditions with carbonyl compounds to give bicyclic oxazolidine products in good overall yields. Moreover, an intramolecular alkylation/desilylation/cycloaddition sequence with the N-trimethylsilylmethyl-1,2-dihydropyridines affords tricyclic indolizidines that incorporate quaternary carbons and up to five stereocenters with good to excellent regio- and diastereoselectivity.
Co-reporter:Jeffrey A. Boerth and Jonathan A. Ellman
Chemical Science 2016 vol. 7(Issue 2) pp:1474-1479
Publication Date(Web):01 Dec 2015
DOI:10.1039/C5SC04138D
The Rh(III)-catalyzed cascade addition of a C–H bond across alkene and carbonyl π-bonds is reported. The reaction proceeds under mild reaction conditions with low catalyst loading. A range of directing groups were shown to be effective as was the functionalization of alkenyl in addition to aromatic C(sp2)–H bonds. When the enone and aldehyde electrophile were tethered together, cyclic β-hydroxy ketones with three contiguous stereocenters were obtained with high diastereoselectivity. The intermolecular three-component cascade reaction was demonstrated for both aldehyde and imine electrophiles. Moreover, the first X-ray structure of a cationic Cp*Rh(III) enolate with interatomic distances consistent with an η3-bound enolate is reported.
Co-reporter:Adam B. Weinstein and Jonathan A. Ellman
Organic Letters 2016 Volume 18(Issue 13) pp:3294-3297
Publication Date(Web):June 23, 2016
DOI:10.1021/acs.orglett.6b01611
The development of Rh(III)-catalyzed C–H conjugate addition/cyclization reactions that provide access to synthetically useful fused bi- and tricyclic nitrogen heterocycles is reported. A broad scope of C–H functionalization substrates and electrophilic olefin coupling partners is effective, and depending on the nature of the directing group, cyclic imide, amide, or heteroaromatic products are obtained. An efficient synthesis of a pyrrolophenanthridine alkaloid natural product, oxoassoanine, highlights the utility of this method.
Co-reporter:Tyler J. Potter and Jonathan A. Ellman
Organic Letters 2016 Volume 18(Issue 15) pp:3838-3841
Publication Date(Web):July 20, 2016
DOI:10.1021/acs.orglett.6b01846
A Rh(III)-catalyzed C–H bond addition/primary amine-promoted cyclization of bis-Michael acceptors is reported. The C–H bond addition step occurs with high chemoselectivity, and the subsequent intramolecular Michael addition, mediated by a primary amine catalyst, sets three contiguous stereocenters with high diastereoselectivity. A broad range of directing groups and both aromatic and alkenyl C–H bonds were shown to be effective in this transformation, affording functionalized piperidines, tetrahydropyrans, and cyclohexanes.
Co-reporter:James P. Phelan
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 11) pp:1713-1718
Publication Date(Web):
DOI:10.1002/adsc.201600110
Co-reporter:Haya Jamali, Hasan A. Khan, Caroline C. Tjin, and Jonathan A. Ellman
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 9) pp:847
Publication Date(Web):July 20, 2016
DOI:10.1021/acsmedchemlett.6b00215
The protein arginine deiminases (PADs) catalyze the post-translational deimination of arginine side chains. Multiple PAD isozymes have been characterized, and abnormal PAD activity has been associated with several human disease states. PAD3 has been characterized as a modulator of cell growth via apoptosis inducing factor and has been implicated in the neurodegenerative response to spinal cord injury. Here, we describe the design, synthesis, and evaluation of conformationally constrained versions of the potent and selective PAD3 inhibitor 2. The cell activity of representative inhibitors in this series was also demonstrated for the first time by rescue of thapsigargin-induced cell death in PAD3-expressing HEK293T cells.Keywords: apoptosis inducing factor; Protein arginine deiminases; small molecule inhibitor; spinal cord injury;
Co-reporter:Apiwat Wangweerawong, Joshua R. Hummel, Robert G. Bergman, and Jonathan A. Ellman
The Journal of Organic Chemistry 2016 Volume 81(Issue 4) pp:1547-1557
Publication Date(Web):February 4, 2016
DOI:10.1021/acs.joc.5b02700
A high yielding and practical two-step synthesis of enantiomerically pure perfluorobutanesulfinamide from Senanayake’s 2-aminoindanol-derived sulfinyl transfer reagent was developed and carried out on a multigram scale. Straightforward condensation of this sulfinamide with ethyl glyoxylate provided the N-perfluorobutanesulfinyl imino ester. The utility of this activated N-sulfinyl imino ester was demonstrated for reactions that gave either no product or very low yields with the corresponding less electrophilic N-tert-butanesulfinyl derivative. Specifically, the Rh(III)-catalyzed C–H bond addition of aromatic compounds to the N-perfluorobutanesulfinyl imino ester provided arylglycines with very high diastereoselectivities for a range of directing groups including pyrrolidine amide, azo, sulfoximine, 1-pyrazole, and 1,2,3-triazole functionalities. Thermal asymmetric aza-Diels–Alder reactions also proceeded in good yields and with high selectivity, including for the substituted dienes (E)-1,3-pentadiene and (2E,4E)-2,4-hexadiene.
Co-reporter:Haya Jamali; Hasan A. Khan; Joseph R. Stringer; Somenath Chowdhury
Journal of the American Chemical Society 2015 Volume 137(Issue 10) pp:3616-3621
Publication Date(Web):March 5, 2015
DOI:10.1021/jacs.5b00095
The protein arginine deiminases (PADs) are a family of enzymes that catalyze the post-translational hydrolytic deimination of arginine residues. Four different enzymologically active PAD subtypes have been characterized and exhibit tissue-specific expression and association with a number of different diseases. In this Article we describe the development of an approach for the reliable discovery of low molecular weight, nonpeptidic fragment substrates of the PADs that then can be optimized and converted to mechanism-based irreversible PAD inhibitors. The approach is demonstrated by the development of potent and selective inhibitors of PAD3, a PAD subtype implicated in the neurodegenerative response to spinal cord injury. Multiple structurally distinct inhibitors were identified with the most potent inhibitors having >10,000 min–1 M–1 kinact/KI values and ≥10-fold selectivity for PAD3 over PADs 1, 2, and 4.
Co-reporter:Shuming Chen, Robert G. Bergman, and Jonathan A. Ellman
Organic Letters 2015 Volume 17(Issue 11) pp:2567-2569
Publication Date(Web):May 20, 2015
DOI:10.1021/acs.orglett.5b00979
A Rh(III)-catalyzed C–H functionalization approach was developed for the preparation of multisubstituted 3-fluoropyridines from α-fluoro-α,β-unsaturated oximes and alkynes. Oximes substituted with aryl, heteroaryl, and alkyl β-substituents were effective coupling partners, as were symmetrical and unsymmetrical alkynes with aryl and alkyl substituents. The first examples of coupling α,β-unsaturated oximes with terminal alkynes was also demonstrated and proceeded with uniformly high regioselectivity to provide single 3-fluoropyridine regioisomers. Reactions were also conveniently set up in air on the benchtop.
Co-reporter:Joshua R. Hummel and Jonathan A. Ellman
Organic Letters 2015 Volume 17(Issue 10) pp:2400-2403
Publication Date(Web):May 6, 2015
DOI:10.1021/acs.orglett.5b00910
The first examples of cobalt(III)-catalyzed C–H bond addition to isocyanates are described, providing a convergent strategy for arene and heteroarene amidation. Using a robust air- and moisture-stable catalyst, this transformation demonstrates a broad isocyanate scope and good functional-group compatibility and has been performed on gram scale.
Co-reporter:Kate D. Otley and Jonathan A. Ellman
Organic Letters 2015 Volume 17(Issue 5) pp:1332-1335
Publication Date(Web):February 25, 2015
DOI:10.1021/acs.orglett.5b00340
The development of a method for the Rh(III)-catalyzed direct vinylation of an aromatic C–H bond to give functionalized styrenes in good yield, using vinyl acetate as a convenient and inexpensive vinyl source, is reported. High functional group tolerance is demonstrated for electronically distinct arenes as well as different directing groups. Mechanistic investigation resulted in the characterization of a novel rhodium–metallacycle, which represents the first X-ray structure of a [1,2]-Rh(III)-alkenyl addition adduct.
Co-reporter:Tyler D. Baguley, Angus C. Nairn, Paul J. Lombroso, Jonathan A. Ellman
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 5) pp:1044-1046
Publication Date(Web):1 March 2015
DOI:10.1016/j.bmcl.2015.01.020
Striatal-enriched protein tyrosine phosphatase (STEP) is a brain specific protein tyrosine phosphatase that has been implicated in many neurodegenerative diseases, such as Alzheimer’s disease. We recently reported the benzopentathiepin TC-2153 as a potent inhibitor of STEP in vitro, cells and animals. Herein, we report the synthesis and evaluation of TC-2153 analogs in order to define what structural features are important for inhibition and to identify positions tolerant of substitution for further study. The trifluoromethyl substitution is beneficial for inhibitor potency, and the amine is tolerant of acylation, and thus provides a convenient handle for introducing additional functionality such as reporter groups.
Co-reporter:Shuming Chen, Brandon Q. Mercado, Robert G. Bergman, and Jonathan A. Ellman
The Journal of Organic Chemistry 2015 Volume 80(Issue 13) pp:6660-6668
Publication Date(Web):June 22, 2015
DOI:10.1021/acs.joc.5b00816
Diastereoselective epoxidation and regioselective ring-opening methods were developed for the synthesis of densely substituted, oxygenated piperidines from two classes of tetrahydropyridines with distinct stereochemical displays of functionalities. A new and practical in situ prepared epoxidation reagent was developed for the diastereoselective epoxidation of one class of sterically hindered tetrahydropyridines. The novel bifunctional epoxidation reagent, 2-carboperoxy-3,4,5,6-tetrafluorobenzoic acid, was designed to incorporate highly reactive percarboxy acid and pendant carboxylic acid groups, which through hydrogen bonding to the amino group successfully overrode steric effects and directed epoxidation to occur at the more hindered face of the tetrahydropyridine. Nucleophilic ring-opening of the epoxides with water, alcohols, and HF proceeded with high regioselectivity, affording piperidinol products with adjacent tetrasubstituted carbons.
Co-reporter:Apiwat Wangweerawong ; Robert G. Bergman
Journal of the American Chemical Society 2014 Volume 136(Issue 24) pp:8520-8523
Publication Date(Web):June 5, 2014
DOI:10.1021/ja5033452
The first asymmetric intermolecular addition of non-acidic C–H bonds to imines is reported. The use of the activating N-perfluorobutanesulfinyl imine substituent is essential for achieving sufficient reactivity and provides outstanding diastereoselectivity (>98:2 dr). Straightforward removal of the sulfinyl group with HCl yields the highly enantiomerically enriched amine hydrochlorides.
Co-reporter:Joshua R. Hummel
Journal of the American Chemical Society 2014 Volume 137(Issue 1) pp:490-498
Publication Date(Web):December 10, 2014
DOI:10.1021/ja5116452
The development of operationally straightforward and cost-effective routes for the assembly of heterocycles from simple inputs is important for many scientific endeavors, including pharmaceutical, agrochemical, and materials research. In this article we describe the development of a new air-stable cationic Co(III) catalyst for convergent, one-step benchtop syntheses of N-aryl-2H-indazoles and furans by C–H bond additions to aldehydes followed by in situ cyclization and aromatization. Only a substoichiometric amount of AcOH is required as an additive that is both low-cost and convenient to handle. The syntheses of these heterocycles are the first examples of Co(III)-catalyzed additions to aldehydes, and reactions are demonstrated for a variety of aromatic, heteroaromatic, and aliphatic derivatives. The syntheses of both N-aryl-2H-indazoles and furans have been performed on 20 mmol scales and should be readily applicable to larger scales. The reported heterocycle syntheses also demonstrate the use of directing groups that have not previously been applied to Co(III)-catalyzed C–H bond functionalizations. Additionally, the synthesis of furans demonstrates the first example of Co(III)-catalyzed functionalization of alkenyl C–H bonds.
Co-reporter:Andrew W. Buesking and Jonathan A. Ellman
Chemical Science 2014 vol. 5(Issue 5) pp:1983-1987
Publication Date(Web):07 Mar 2014
DOI:10.1039/C4SC00084F
We describe the Rh-catalyzed addition of α-sulfinamido trifluoroborates to carbonyl compounds for the convergent, asymmetric synthesis of vicinal amino alcohols. This method represents the first application of α-amino boron reagents as reaction partners in rhodium-catalyzed couplings. Reactions with trifluoromethyl ketones proceed in reasonable yields and with good diastereoselectivity along with complete retention at the organoboron stereocenter. The potential of this method is further highlighted by the exploration of a variety of nitrogen substituents and addition to benzaldehyde and trityl-protected isatin.
Co-reporter:Tehetena Mesganaw and Jonathan A. Ellman
Organic Process Research & Development 2014 Volume 18(Issue 9) pp:1097-1104
Publication Date(Web):August 29, 2014
DOI:10.1021/op500224x
A Rh-catalyzed C–H bond activation/alkenylation/electrocyclization cascade reaction provides diverse 1,2-dihydropyridines from simple and readily available precursors. The reaction can be carried out at low (<1%) Rh-catalyst loadings, and the use of the robust, air-stable Rh precatalyst, [RhCl(cod)]2, enables the cascade reaction to be easily performed on the benchtop. The 1,2-dihydropyridine products serve as extremely versatile synthetic intermediates for further elaboration often without isolation. The addition of electrophiles under kinetic or thermodynamic conditions provides a wide range of iminiums. Subsequent addition of a nucleophile then generates a diverse array of differently substituted piperidine products. Additionally, [3 + 2] and [4 + 2] cycloadditions of the 1,2-dihydropyridine intermediate provides access to bridged bicyclic structures such as tropanes and isoquinuclidines. These concise reaction sequences enable the formation of highly substituted piperidines in synthetically useful yields with excellent diastereoselectivity.
Co-reporter:Tehetena Mesganaw and Jonathan A. Ellman
Organic Process Research & Development 2014 Volume 18(Issue 9) pp:1105-1109
Publication Date(Web):August 29, 2014
DOI:10.1021/op500225c
We report a Rh(I)-catalyzed C–H activation/alkenylation/electrocyclization cascade and subsequent reduction for the synthesis of highly substituted tetrahydropyridines. These products can be accessed on a gram scale with low catalyst loadings and at high reaction concentrations. Additionally, a modified Rh-catalyst, prepared from [RhCl(cod)]2 as a robust bench-stable precatalyst was developed to enable straightforward reaction set up without the use of a glovebox. To demonstrate the practicality of this reaction, a >100 mmol scale Rh-catalyzed cascade reaction sequence utilizing the air-stable precatalyst [RhCl(cod)]2 was performed on the bench to furnish the pure tetrahydropyridine product in 93% yield.
Co-reporter:Simon Duttwyler;Shuming Chen;Colin Lu;Bron Q. Mercado;Dr. Robert G. Bergman;Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2014 Volume 53( Issue 15) pp:3877-3880
Publication Date(Web):
DOI:10.1002/anie.201310517
Abstract
The first example of C alkylation of 1,2-dihydropyridines with alkyl triflates and Michael acceptors was developed to introduce quaternary carbon centers with high regio- and diastereoselectivity. Hydride or carbon nucleophile addition to the resultant iminium ion also proceeded with high diastereoselectivity. Carbon nucleophile addition results in an unprecedented level of substitution to provide piperidine rings with adjacent tetrasubstituted carbon atoms.
Co-reporter:James P. Phelan;Evan J. Patel ;Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2014 Volume 53( Issue 42) pp:11329-11332
Publication Date(Web):
DOI:10.1002/anie.201406971
Abstract
The first example of a catalytic enantioselective addition to and nitronate protonation of trisubstituted nitroalkenes to produce highly enantioenriched products with a tetrasubstituted carbon is reported. Thioacids added in excellent yields and with high enantioselectivities to both activated and unactivated nitroalkenes. The 1,2-nitrothioacetate products can be readily converted in two steps to biomedically relevant 1,2-aminosulfonic acids without loss of enantiopurity.
Co-reporter:James P. Phelan;Evan J. Patel ;Dr. Jonathan A. Ellman
Angewandte Chemie 2014 Volume 126( Issue 42) pp:11511-11514
Publication Date(Web):
DOI:10.1002/ange.201406971
Abstract
The first example of a catalytic enantioselective addition to and nitronate protonation of trisubstituted nitroalkenes to produce highly enantioenriched products with a tetrasubstituted carbon is reported. Thioacids added in excellent yields and with high enantioselectivities to both activated and unactivated nitroalkenes. The 1,2-nitrothioacetate products can be readily converted in two steps to biomedically relevant 1,2-aminosulfonic acids without loss of enantiopurity.
Co-reporter:Kate D. Otley and Jonathan A. Ellman
The Journal of Organic Chemistry 2014 Volume 79(Issue 17) pp:8296-8303
Publication Date(Web):August 26, 2014
DOI:10.1021/jo5015432
We report here a new, atom economical annulation to 2,1-benzisoxazole scaffolds via the BF3·Et2O-catalyzed reaction of glyoxylate esters and nitrosoarenes. The developed method represents a convergent route to this compound class from previously unexplored inputs and provides a range of 2,1-benzisoxazoles in moderate to high yields under convenient conditions. Along with exploration of substrate scope, initial mechanistic investigation through 18O labeling and the synthesis of a reaction intermediate provides evidence for an unusual umpolung addition of glyoxylates to nitrosobenzenes with high O-selectivity, followed by a new type of Friedel–Crafts cyclization.
Co-reporter:Andrew W. Buesking, Vlad Bacauanu, Irene Cai, and Jonathan A. Ellman
The Journal of Organic Chemistry 2014 Volume 79(Issue 8) pp:3671-3677
Publication Date(Web):March 31, 2014
DOI:10.1021/jo500300t
The asymmetric borylation of N-tert-butanesulfinyl imines with bis(pinacolato)diboron is achieved using a Cu(II) catalyst and provides access to synthetically useful and pharmaceutically relevant α-amino boronic acid derivatives. The Cu(II)-catalyzed reaction is performed on the benchtop in air at room temperature using commercially available, inexpensive reagents at low catalyst loadings. A variety of N-tert-butanesulfinyl imines, including ketimines, react readily to provide α-sulfinamido boronate esters in good yields and with high stereoselectivity. In addition, this transformation is applied to the straightforward, telescoped synthesis of α-sulfinamido trifluoroborates.
Co-reporter:Simon Duttwyler;Shuming Chen;Colin Lu;Bron Q. Mercado;Dr. Robert G. Bergman;Dr. Jonathan A. Ellman
Angewandte Chemie 2014 Volume 126( Issue 15) pp:3958-3961
Publication Date(Web):
DOI:10.1002/ange.201310517
Abstract
The first example of C alkylation of 1,2-dihydropyridines with alkyl triflates and Michael acceptors was developed to introduce quaternary carbon centers with high regio- and diastereoselectivity. Hydride or carbon nucleophile addition to the resultant iminium ion also proceeded with high diastereoselectivity. Carbon nucleophile addition results in an unprecedented level of substitution to provide piperidine rings with adjacent tetrasubstituted carbon atoms.
Co-reporter:Yajing Lian ; Robert G. Bergman ; Luke D. Lavis
Journal of the American Chemical Society 2013 Volume 135(Issue 19) pp:7122-7125
Publication Date(Web):May 3, 2013
DOI:10.1021/ja402761p
An efficient, one-step, and highly functional group-compatible synthesis of substituted N-aryl-2H-indazoles is reported via the rhodium(III)-catalyzed C–H bond addition of azobenzenes to aldehydes. The regioselective coupling of unsymmetrical azobenzenes was further demonstrated and led to the development of a new removable aryl group that allows for the preparation of indazoles without N-substitution. The 2-aryl-2H-indazole products also represent a new class of readily prepared fluorophores for which initial spectroscopic characterization has been performed.
Co-reporter:Mikaël Brasse ; Juan Cámpora ; Jonathan A. Ellman ;Robert G. Bergman
Journal of the American Chemical Society 2013 Volume 135(Issue 17) pp:6427-6430
Publication Date(Web):April 5, 2013
DOI:10.1021/ja401561q
The Rh(III)-catalyzed oxidative coupling of alkenes with arenes provides a greener alternative to the classical Heck reaction for the synthesis of arene-functionalized alkenes. The present mechanistic study gives insights for the rational development of this key transformation. The catalyst resting states and the rate law of the reaction have been identified. The reaction rate is solely dependent on the catalyst and alkene concentrations, and the turnover-limiting step is the migratory insertion of the alkene into a Rh–C(aryl) bond.
Co-reporter:Michael A. Ischay ; Michael K. Takase ; Robert G. Bergman
Journal of the American Chemical Society 2013 Volume 135(Issue 7) pp:2478-2481
Publication Date(Web):February 11, 2013
DOI:10.1021/ja312311k
Acid treatment of densely substituted 2-silyl-1,2-dihydropyridines provides a new and convenient entry to reactive azomethine ylides that can (1) be protonated and reduced with high stereoselectivity to give piperidines, (2) participate in [3 + 2] dipolar cycloaddition to give tropanes, and (3) undergo a Nazarov-like 6-π electrocyclization that upon reduction give 2-azabicyclo[3.1.0] systems.
Co-reporter:Rhia M. Martin, Robert G. Bergman, and Jonathan A. Ellman
Organic Letters 2013 Volume 15(Issue 3) pp:444-447
Publication Date(Web):January 15, 2013
DOI:10.1021/ol303040r
A stereo- and regioselective Diels–Alder reaction for the synthesis of highly substituted isoquinuclidines from dihydropyridines and electron-deficient alkenes has been developed. While reactions with activated dienophiles proceed readily under thermal conditions, the use of Lewis acid additives is necessary to facilitate cycloadditions for less reactive alkenes. This procedure affords the target compounds in high yields and diastereoselectivities.
Co-reporter:Tyler D. Baguley ; Hai-Chao Xu ; Manavi Chatterjee ; Angus C. Nairn ; Paul J. Lombroso
Journal of Medicinal Chemistry 2013 Volume 56(Issue 19) pp:7636-7650
Publication Date(Web):October 1, 2013
DOI:10.1021/jm401037h
High levels of striatal-enriched protein tyrosine phosphatase (STEP) activity are observed in a number of neuropsychiatric disorders such as Alzheimer’s disease. Overexpression of STEP results in the dephosphorylation and inactivation of many key neuronal signaling molecules, including ionotropic glutamate receptors. Moreover, genetically reducing STEP levels in AD mouse models significantly reversed cognitive deficits and decreased glutamate receptor internalization. These results support STEP as a potential target for drug discovery for the treatment of Alzheimer’s disease. Herein, a substrate-based approach for the discovery and optimization of fragments called substrate activity screening (SAS) has been applied to the development of low molecular weight (<450 Da) and nonpeptidic, single-digit micromolar mechanism-based STEP inhibitors with greater than 20-fold selectivity across multiple tyrosine and dual specificity phosphatases. Significant levels of STEP inhibition in rat cortical neurons are also observed.
Co-reporter:Yajing Lian;Tatjana Huber;Kevin D. Hesp;Dr. Robert G. Bergman;Dr. Jonathan A. Ellman
Angewandte Chemie International Edition 2013 Volume 52( Issue 2) pp:629-633
Publication Date(Web):
DOI:10.1002/anie.201207995
Co-reporter:Yajing Lian;Tatjana Huber;Kevin D. Hesp;Dr. Robert G. Bergman;Dr. Jonathan A. Ellman
Angewandte Chemie 2013 Volume 125( Issue 2) pp:657-661
Publication Date(Web):
DOI:10.1002/ange.201207995
Co-reporter:Robert G. Bergman;Kenneth B. Wiberg;Simon Duttwyler;Michael K. Takase;Shuming Chen
Science 2013 Volume 339(Issue 6120) pp:678-682
Publication Date(Web):08 Feb 2013
DOI:10.1126/science.1230704
Co-reporter:Denise A. Colby, Andy S. Tsai, Robert G. Bergman, and Jonathan A. Ellman
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:814
Publication Date(Web):December 8, 2011
DOI:10.1021/ar200190g
Over the last several decades, researchers have achieved remarkable progress in the field of organometallic chemistry. The development of metal-catalyzed cross-coupling reactions represents a paradigm shift in chemical synthesis, and today synthetic chemists can readily access carbon–carbon and carbon–heteroatom bonds from a vast array of starting compounds. Although we cannot understate the importance of these methods, the required prefunctionalization to carry out these reactions adds cost and reduces the availability of the starting reagents.The use of C–H bond activation in lieu of prefunctionalization has presented a tantalizing alternative to classical cross-coupling reactions. Researchers have met the challenges of selectivity and reactivity associated with the development of C–H bond functionalization reactions with an explosion of creative advances in substrate and catalyst design. Literature reports on selectivity based on steric effects, acidity, and electronic and directing group effects are now numerous.Our group has developed an array of C–H bond functionalization reactions that take advantage of a chelating directing group, and this Account surveys our progress in this area. The use of chelation control in C–H bond functionalization offers several advantages with respect to substrate scope and application to total synthesis. The predictability and decreased dependence on the inherent stereoelectronics of the substrate generally result in selective and high yielding transformations with broad applicability. The nature of the chelating moiety can be chosen to serve as a functional handle in subsequent elaborations.Our work began with the use of Rh(I) catalysts in intramolecular aromatic C–H annulations, which we further developed to include enantioselective transformations. The application of this chemistry to the simple olefinic C–H bonds found in α,β-unsaturated imines allowed access to highly substituted olefins, pyridines, and piperidines. We observed complementary reactivity with Rh(III) catalysts and developed an oxidative coupling with unactivated alkenes. Further studies on the Rh(III) catalysts led us to develop methods for the coupling of C–H bonds to polarized π bonds such as those in imines and isocyanates. In several cases the methods that we have developed for chelation-controlled C–H bond functionalization have been applied to the total synthesis of complex molecules such as natural products, highlighting the utility of these methods in organic synthesis.
Co-reporter:Kyle L. Kimmel ; Jimmie D. Weaver ; Melissa Lee
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9058-9061
Publication Date(Web):May 23, 2012
DOI:10.1021/ja3026196
The highly enantioselective protonation of nitronates formed upon the addition of α-substituted Meldrum’s acids to terminally unsubstituted nitroalkenes is described. This work represents the first enantioselective catalytic addition of any type of nucleophile to this class of nitroalkenes. Moreover, for the successful implementation of this method, a new type of N-sulfinyl urea catalyst with chirality residing only at the sulfinyl group was developed, thereby enabling the incorporation of a diverse range of achiral diamine motifs. Finally, the Meldrum’s acid addition products were readily converted to pharmaceutically relevant 3,5-disubstituted pyrrolidinones in high yield.
Co-reporter:Michael E. Tauchert ; Christopher D. Incarvito ; Arnold L. Rheingold ; Robert G. Bergman
Journal of the American Chemical Society 2012 Volume 134(Issue 3) pp:1482-1485
Publication Date(Web):January 17, 2012
DOI:10.1021/ja211110h
Rh(III)-catalyzed arylation of imines provides a new method for C–C bond formation while simultaneously introducing an α-branched amine as a functional group. This detailed mechanistic study provides insights for the rational future development of this new reaction. Relevant intermediate Rh(III) complexes have been isolated and characterized, and their reactivities in stoichiometric reactions with relevant substrates have been monitored. The reaction was found to be first order in the catalyst resting state and inverse first order in the C–H activation substrate.
Co-reporter:Simon Duttwyler ; Colin Lu ; Arnold L. Rheingold ; Robert G. Bergman
Journal of the American Chemical Society 2012 Volume 134(Issue 9) pp:4064-4067
Publication Date(Web):February 22, 2012
DOI:10.1021/ja2119833
A versatile reaction cascade leading to highly substituted 1,2,3,6-tetrahydropyridines has been developed. It comprises rhodium(I)-catalyzed C–H activation–alkyne coupling followed by electrocyclization and subsequent acid/borohydride-promoted reduction. This one-pot procedure affords the target compounds in up to 95% yield with >95% diastereomeric purity.
Co-reporter:Yajing Lian, Robert G. Bergman and Jonathan A. Ellman
Chemical Science 2012 vol. 3(Issue 10) pp:3088-3092
Publication Date(Web):02 Aug 2012
DOI:10.1039/C2SC20835K
We herein report the Rh(III)-catalyzed C–H bond activation and addition of benzimidates to aldehydes to afford biologically important phthalides in a single step. The imidate is a novel and unexplored directing group that not only enables C–H bond activation and addition to aldehydes, but also serves to capture the reversibly formed alcohol intermediate. The reaction shows broad scope with a high level of functional group compatibility and is applicable to both aromatic and aliphatic aldehydes.
Co-reporter:Kyle L. Kimmel, Jimmie D. Weaver and Jonathan A. Ellman
Chemical Science 2012 vol. 3(Issue 1) pp:121-125
Publication Date(Web):25 Aug 2011
DOI:10.1039/C1SC00441G
Using N-sulfinyl urea catalysis, a method has been developed for the asymmetric synthesis of biologically important γ-amino acids with a high level of efficiency, practicality and unprecedented control of multiple stereocenters. This method is based upon the highly enantio- and diastereoselective addition of cyclohexyl Meldrum's acid as an easily deprotectable monocarboxylic acid equivalent. The addition to both β-substituted and α,β-disubstituted nitroalkenes using N-sulfinyl urea organocatalyst 8 is described. The utility of this new method toward drug production is demonstrated by the mole scale preparation of a key precursor to the commercial drug Lyrica using catalyst 8 at only 0.2 mol% loading. Moreover, α,β-disubstituted nitroalkene addition products were efficiently converted to γ-amino acid derivatives without epimerization of either stereocenter.
Co-reporter:Kevin D. Hesp, Robert G. Bergman, and Jonathan A. Ellman
Organic Letters 2012 Volume 14(Issue 9) pp:2304-2307
Publication Date(Web):April 13, 2012
DOI:10.1021/ol300723x
Rhodium-catalyzed addition of benzamide C–H bonds to a range of aromatic N-sulfonyl aldimines has been developed and proceeds with high functional group compatibility. The synthetic utility of the resulting branched amine products has also been demonstrated by the preparation of isoindoline and isoindolinone frameworks.
Co-reporter:Kyle L. Kimmel, MaryAnn T. Robak, Stephen Thomas, Melissa Lee, Jonathan A. Ellman
Tetrahedron 2012 68(12) pp: 2704-2712
Publication Date(Web):
DOI:10.1016/j.tet.2012.01.048
Co-reporter:Hyung Hoon Jung, Andrew W. Buesking, and Jonathan A. Ellman
The Journal of Organic Chemistry 2012 Volume 77(Issue 21) pp:9593-9600
Publication Date(Web):October 24, 2012
DOI:10.1021/jo301634y
Arylboroxines, which are easily accessed by drying commercially available arylboronic acids, are added to N-(isopropanesulfinyl)ketimines derived from cyclohexanone, N-Boc-piperidin-4-one, and tetrahydropyran-4-one in high yields and with excellent functional group compatibility via rhodium catalysis. These results contrast with additions to the corresponding ketimines incorporating the larger N-tert-butanesulfinyl group, which give considerably lower yields. Efficient two-step preparation of racemic isopropanesulfinamide from inexpensive isopropyl disulfide and recycling of the isopropanesulfinyl group from the addition products are also described.
Co-reporter:Rhia M. Martin, Robert G. Bergman, and Jonathan A. Ellman
The Journal of Organic Chemistry 2012 Volume 77(Issue 5) pp:2501-2507
Publication Date(Web):February 14, 2012
DOI:10.1021/jo202280e
An expedient one-pot rhodium catalyzed C–H bond functionalization/electrocyclization/dehydration procedure has been developed for the synthesis of highly substituted pyridine derivatives from terminal alkynes and α,β-unsaturated ketoximes. The use of electron-deficient phosphite ligands is important to suppress dimerization of the terminal alkynes to enynes.
Co-reporter:Kevin D. Hesp ; Robert G. Bergman
Journal of the American Chemical Society 2011 Volume 133(Issue 30) pp:11430-11433
Publication Date(Web):June 29, 2011
DOI:10.1021/ja203495c
A Rh(III)-catalyzed protocol for the amidation of anilide and enamide C–H bonds with isocyanates has been developed. This method provides direct and efficient syntheses of N-acyl anthranilamides, enamine amides, and pyrimidin-4-one heterocycles.
Co-reporter:Andy S. Tsai ; Michael E. Tauchert ; Robert G. Bergman
Journal of the American Chemical Society 2011 Volume 133(Issue 5) pp:1248-1250
Publication Date(Web):January 4, 2011
DOI:10.1021/ja109562x
The first rhodium-catalyzed arylation of imines proceeding via C−H bond functionalization is reported. Use of a non-coordinating halide abstractor is important to obtain reactivity. Aryl-branched N-Boc-amines are formed, and a wide range of functionality is compatible with the reaction.
Co-reporter:Andrew W. Buesking, Tyler D. Baguley, and Jonathan A. Ellman
Organic Letters 2011 Volume 13(Issue 5) pp:964-967
Publication Date(Web):February 9, 2011
DOI:10.1021/ol103002t
The MgCl2-enhanced addition of benzyl zinc reagents to N-tert-butanesulfinyl imines proceeds readily at room temperature to afford the N-tert-butanesulfinyl-protected amine products in good yields and diastereomeric ratios. This method is functional group tolerant in both the imine substrate and benzyl zinc coupling partner. Moreover, benzyl zinc reagent addition to the N-tert-butanesulfinyl imine 3o prepared from isopropylidene-protected glyceraldehyde proceeds in high yield and with exceptional selectivity to provide rapid entry to hydroxyethylamine-based aspartyl protease inhibitors.
Co-reporter:Andy S. Tsai, Mikaël Brasse, Robert G. Bergman, and Jonathan A. Ellman
Organic Letters 2011 Volume 13(Issue 3) pp:540-542
Publication Date(Web):December 22, 2010
DOI:10.1021/ol102890k
Oxime directed aromatic C−H bond activation and oxidative coupling to alkenes is reported using a cationic Rh(III) catalyst. Significantly, the method can be used to oxidatively couple unactivated, aliphatic alkenes.
Co-reporter:Hyung Hoon Jung, Andrew W. Buesking, and Jonathan A. Ellman
Organic Letters 2011 Volume 13(Issue 15) pp:3912-3915
Publication Date(Web):July 6, 2011
DOI:10.1021/ol201438k
The rhodium-catalyzed addition of readily accessible arylboroxines to N-tert-butanesulfinyl ketimines derived from oxetan-3-one, N-Boc-azetidin-3-one, and isatins proceeds in high yields with excellent functional group compatibility. Moreover, high diastereoselectivities are observed for the additions to the N-sulfinyl ketimines derived from isatins.
Co-reporter:Mikaël Brasse, Jonathan A. Ellman and Robert G. Bergman
Chemical Communications 2011 vol. 47(Issue 17) pp:5019-5021
Publication Date(Web):29 Mar 2011
DOI:10.1039/C1CC10507H
A simple, solvent-free, one-pot autoxidative coupling reaction between quinoline and indoles or pyrroles is reported. This atom economic method requires only a stoichiometric amount of inexpensive hydrochloric acid and does not require a catalyst.
Co-reporter:Mark R. Crimmin, Denise A. Colby, Jonathan A. Ellman and Robert G. Bergman
Dalton Transactions 2011 vol. 40(Issue 2) pp:514-522
Publication Date(Web):25 Nov 2010
DOI:10.1039/C0DT01267J
A series of N1,N1,N3-tri-substituted benzamidrazones of the general formula [PhC(NHR)NNMe2] (R = Me, n-Pr, i-Pr, n-Bu, Bn, Ph; 1a–f) was synthesized via condensation of 1,1-dimethylhydrazine with the corresponding imidoyl chloride, [PhC(Cl)NR]. Multinuclear NMR data, and zero-point energy DFT calculations conducted with the B3LYP functional and 6–31G+(d,p) basis set, suggest that these compounds exist as a single tautomer in solution; possessing a weak intramolecular hydrogen bond and a structure dominated by the localised resonance structure ArC(NHR)N–NMe2. An X-ray crystallographic study upon PhC(NHPh)NNMe2 (1f) demonstrated that this compound adopts an identical tautomer in the solid state. Reactions of [PhC(NHMe)NNMe2] (1a) with [LMCl2]2 (M = Ru, L = cymene; M = Rh, Ir, L = Cp*) results in the stoichiometric formation of products of the formula [LM{PhC(NMe)NHNMe2}Cl]+Cl− (2a–c) in which the amidrazone chelates the metal in a κ2-N1,N3-coordination mode. Formation of this five-membered chelate occurs with a concomitant tautomerisation of the amidrazone ligand to an alternative tautomer, i.e. [PhC(NMe)NHNMe2], the latter tautomer is expected to be readily energetically accessible based upon the aforementioned DFT calculations. This series of salts may be deprotonated with lithium hexamethyldisilazide to form the corresponding charge neutral complexes [LM{PhC(NMe)NNMe2}] (3a–c). In contrast, the reaction of N1,N1,N3-tri-substituted benzamidrazones with [(cymene)RuCl2]2 in the presence of NaOAc yielded a mixture of cyclometallation (C–H activation) and amidrazone chelation/deprotonation (N-H activation) products. Reaction of 1a yielded an inseparable mixture of products, whilst the reaction of 1c resulted in formation of the cyclometallated product [LM{C6H5C(NiPr)NHNMe2}] (L = cymene, M = Ru; 4a) in a modest 62% yield. This latter complex could be isolated as a crystalline orange solid, full characterisation including single crystal X-ray diffraction demonstrated that the amidrazone coordinates in a κ2-N2,C-coordination mode.
Co-reporter:William C. Floyd III;Dr. Gopal K. Datta;Dr. Shinichi Imamura;Heidi M. Kieler-Ferguson;Katherine Jerger;Dr. Andrew W. Patterson;Megan E. Fox; Francis C. Szoka; Jean M. J. Fréchet; Jonathan A. Ellman
ChemMedChem 2011 Volume 6( Issue 1) pp:49-53
Publication Date(Web):
DOI:10.1002/cmdc.201000377
Co-reporter:MaryAnn T. Robak, Melissa A. Herbage, Jonathan A. Ellman
Tetrahedron 2011 67(24) pp: 4412-4416
Publication Date(Web):
DOI:10.1016/j.tet.2011.03.030
Co-reporter:Elena Arceo ; Jonathan A. Ellman ;Robert G. Bergman
Journal of the American Chemical Society 2010 Volume 132(Issue 33) pp:11408-11409
Publication Date(Web):July 29, 2010
DOI:10.1021/ja103436v
A new method for the catalytic didehydroxylation of vicinal diols is described. Employing a readily available low-valent rhenium carbonyl complex and a simple alcohol as a reducing agent, both terminal and internal vicinal diols are deoxygenated to olefins in good yield. The optional addition of acid (TsOH, H2SO4) provides access to lower reaction temperatures. This new system enables the transformation of a four-carbon sugar polyol into an oxygen-reduced compound, providing promising evidence for its practical application to produce unsaturated compounds from biomass-derived materials.
Co-reporter:Jason M. Nichols ; Lee M. Bishop ; Robert G. Bergman
Journal of the American Chemical Society 2010 Volume 132(Issue 36) pp:12554-12555
Publication Date(Web):August 23, 2010
DOI:10.1021/ja106101f
A ruthenium-catalyzed, redox neutral C−O bond cleavage of 2-aryloxy-1-arylethanols was developed that yields cleavage products in 62−98% isolated yield. This reaction is applicable to breaking the key ethereal bond found in lignin-related polymers. The bond transformation proceeds by a tandem dehydrogenation/reductive ether cleavage. Initial mechanistic investigations indicate that the ether cleavage is most likely an organometallic C−O activation. A catalytic depolymerization of a lignin-related polymer quantitatively yields the corresponding monomer with no added reagent.
Co-reporter:Melissa J. Leyva, Francesco DeGiacomo, Linda S. Kaltenbach, Jennifer Holcomb, Ningzhe Zhang, Juliette Gafni, Hyunsun Park, Donald C. Lo, Guy S. Salvesen, Lisa M. Ellerby, Jonathan A. Ellman
Chemistry & Biology 2010 Volume 17(Issue 11) pp:1189-1200
Publication Date(Web):24 November 2010
DOI:10.1016/j.chembiol.2010.08.014
Huntington's Disease (HD) is characterized by a mutation in the huntingtin (Htt) gene encoding an expansion of glutamine repeats on the N terminus of the Htt protein. Numerous studies have identified Htt proteolysis as a critical pathological event in HD postmortem human tissue and mouse HD models, and proteases known as caspases have emerged as attractive HD therapeutic targets. We report the use of the substrate activity screening method against caspase-3 and -6 to identify three novel, pan-caspase inhibitors that block proteolysis of Htt at caspase-3 and -6 cleavage sites. In HD models these irreversible inhibitors suppressed Hdh111Q/111Q-mediated toxicity and rescued rat striatal and cortical neurons from cell death. In this study, the identified nonpeptidic caspase inhibitors were used to confirm the role of caspase-mediated Htt proteolysis in HD. These results further implicate caspases as promising targets for HD therapeutic development.Graphical AbstractFigure optionsDownload full-size imageDownload high-quality image (106 K)Download as PowerPoint slideHighlights► Nonpeptidic caspase inhibitors identified by substrate activity screening method ► Inhibitors block caspase-3 and -6 mediated cleavage of Htt and toxicity in mouse striatal cells ► Inhibitors rescue HttN90Q73-induced degeneration of rat striatal and cortical neurons
Co-reporter:Katherine A. Rawls, Christoph Grundner and Jonathan A. Ellman
Organic & Biomolecular Chemistry 2010 vol. 8(Issue 18) pp:4066-4070
Publication Date(Web):19 Jul 2010
DOI:10.1039/C0OB00182A
The design and synthesis of new inhibitor analogues for the Mycobacterium tuberculosis (Mtb) phosphatase PtpB is described. Analogues were synthesized by incorporation of two common and effective phosphate mimetics, the isothiazolidinone (IZD) and the difluoromethylphosphonic acid (DFMP). The basic scaffold of the inhibitor was identified from structure–activity relationships established for a previously published isoxazole inhibitor, while the phosphate mimetics were chosen based on their proven cell permeability and activity when incorporated into previously reported inhibitors for the phosphatase PTP1B. The inhibitory activity of each compound was evaluated, and each was found to have low or submicromolar affinity for PtpB.
Co-reporter:Ashley M. Berman, Robert G. Bergman, and Jonathan A. Ellman
The Journal of Organic Chemistry 2010 Volume 75(Issue 22) pp:7863-7868
Publication Date(Web):October 29, 2010
DOI:10.1021/jo101793r
The Rh(I)-catalyzed direct arylation of azines has been developed. Quinolines and 2-substituted pyridines couple with aryl bromides to efficiently afford ortho-arylated azine products using the commercially available and air-stable catalyst [RhCl(CO)2]2. Electron-deficient and electron-rich aromatic bromides couple in good yields, and hydroxyl, chloro, fluoro, trifluoromethyl, ether, and ketone functionalities are compatible with the reaction conditions. Aroyl chlorides also serve as effective azine coupling partners to give ortho-arylation products via a decarbonylation pathway.
Co-reporter:Yajing Lian ; Joshua R. Hummel ; Robert G. Bergman
Journal of the American Chemical Society () pp:
Publication Date(Web):August 19, 2013
DOI:10.1021/ja406131a
We report formal [3 + 3] annulations of aromatic azides with aromatic imines and azobenzenes to give acridines and phenazines, respectively. These transformations proceed through a cascade process of Rh(III)-catalyzed amination followed by intramolecular electrophilic aromatic substitution and aromatization. Acridines can be directly prepared from aromatic aldehydes by in situ imine formation using catalytic benzylamine.
Co-reporter:Mikaël Brasse, Jonathan A. Ellman and Robert G. Bergman
Chemical Communications 2011 - vol. 47(Issue 17) pp:NaN5021-5021
Publication Date(Web):2011/03/29
DOI:10.1039/C1CC10507H
A simple, solvent-free, one-pot autoxidative coupling reaction between quinoline and indoles or pyrroles is reported. This atom economic method requires only a stoichiometric amount of inexpensive hydrochloric acid and does not require a catalyst.
Co-reporter:Katherine A. Rawls, Christoph Grundner and Jonathan A. Ellman
Organic & Biomolecular Chemistry 2010 - vol. 8(Issue 18) pp:NaN4070-4070
Publication Date(Web):2010/07/19
DOI:10.1039/C0OB00182A
The design and synthesis of new inhibitor analogues for the Mycobacterium tuberculosis (Mtb) phosphatase PtpB is described. Analogues were synthesized by incorporation of two common and effective phosphate mimetics, the isothiazolidinone (IZD) and the difluoromethylphosphonic acid (DFMP). The basic scaffold of the inhibitor was identified from structure–activity relationships established for a previously published isoxazole inhibitor, while the phosphate mimetics were chosen based on their proven cell permeability and activity when incorporated into previously reported inhibitors for the phosphatase PTP1B. The inhibitory activity of each compound was evaluated, and each was found to have low or submicromolar affinity for PtpB.
Co-reporter:Mark R. Crimmin, Denise A. Colby, Jonathan A. Ellman and Robert G. Bergman
Dalton Transactions 2011 - vol. 40(Issue 2) pp:NaN522-522
Publication Date(Web):2010/11/25
DOI:10.1039/C0DT01267J
A series of N1,N1,N3-tri-substituted benzamidrazones of the general formula [PhC(NHR)NNMe2] (R = Me, n-Pr, i-Pr, n-Bu, Bn, Ph; 1a–f) was synthesized via condensation of 1,1-dimethylhydrazine with the corresponding imidoyl chloride, [PhC(Cl)NR]. Multinuclear NMR data, and zero-point energy DFT calculations conducted with the B3LYP functional and 6–31G+(d,p) basis set, suggest that these compounds exist as a single tautomer in solution; possessing a weak intramolecular hydrogen bond and a structure dominated by the localised resonance structure ArC(NHR)N–NMe2. An X-ray crystallographic study upon PhC(NHPh)NNMe2 (1f) demonstrated that this compound adopts an identical tautomer in the solid state. Reactions of [PhC(NHMe)NNMe2] (1a) with [LMCl2]2 (M = Ru, L = cymene; M = Rh, Ir, L = Cp*) results in the stoichiometric formation of products of the formula [LM{PhC(NMe)NHNMe2}Cl]+Cl− (2a–c) in which the amidrazone chelates the metal in a κ2-N1,N3-coordination mode. Formation of this five-membered chelate occurs with a concomitant tautomerisation of the amidrazone ligand to an alternative tautomer, i.e. [PhC(NMe)NHNMe2], the latter tautomer is expected to be readily energetically accessible based upon the aforementioned DFT calculations. This series of salts may be deprotonated with lithium hexamethyldisilazide to form the corresponding charge neutral complexes [LM{PhC(NMe)NNMe2}] (3a–c). In contrast, the reaction of N1,N1,N3-tri-substituted benzamidrazones with [(cymene)RuCl2]2 in the presence of NaOAc yielded a mixture of cyclometallation (C–H activation) and amidrazone chelation/deprotonation (N-H activation) products. Reaction of 1a yielded an inseparable mixture of products, whilst the reaction of 1c resulted in formation of the cyclometallated product [LM{C6H5C(NiPr)NHNMe2}] (L = cymene, M = Ru; 4a) in a modest 62% yield. This latter complex could be isolated as a crystalline orange solid, full characterisation including single crystal X-ray diffraction demonstrated that the amidrazone coordinates in a κ2-N2,C-coordination mode.
Co-reporter:Kyle L. Kimmel, Jimmie D. Weaver and Jonathan A. Ellman
Chemical Science (2010-Present) 2012 - vol. 3(Issue 1) pp:NaN125-125
Publication Date(Web):2011/08/25
DOI:10.1039/C1SC00441G
Using N-sulfinyl urea catalysis, a method has been developed for the asymmetric synthesis of biologically important γ-amino acids with a high level of efficiency, practicality and unprecedented control of multiple stereocenters. This method is based upon the highly enantio- and diastereoselective addition of cyclohexyl Meldrum's acid as an easily deprotectable monocarboxylic acid equivalent. The addition to both β-substituted and α,β-disubstituted nitroalkenes using N-sulfinyl urea organocatalyst 8 is described. The utility of this new method toward drug production is demonstrated by the mole scale preparation of a key precursor to the commercial drug Lyrica using catalyst 8 at only 0.2 mol% loading. Moreover, α,β-disubstituted nitroalkene addition products were efficiently converted to γ-amino acid derivatives without epimerization of either stereocenter.
Co-reporter:Yajing Lian, Robert G. Bergman and Jonathan A. Ellman
Chemical Science (2010-Present) 2012 - vol. 3(Issue 10) pp:NaN3092-3092
Publication Date(Web):2012/08/02
DOI:10.1039/C2SC20835K
We herein report the Rh(III)-catalyzed C–H bond activation and addition of benzimidates to aldehydes to afford biologically important phthalides in a single step. The imidate is a novel and unexplored directing group that not only enables C–H bond activation and addition to aldehydes, but also serves to capture the reversibly formed alcohol intermediate. The reaction shows broad scope with a high level of functional group compatibility and is applicable to both aromatic and aliphatic aldehydes.
Co-reporter:Andrew W. Buesking and Jonathan A. Ellman
Chemical Science (2010-Present) 2014 - vol. 5(Issue 5) pp:NaN1987-1987
Publication Date(Web):2014/03/07
DOI:10.1039/C4SC00084F
We describe the Rh-catalyzed addition of α-sulfinamido trifluoroborates to carbonyl compounds for the convergent, asymmetric synthesis of vicinal amino alcohols. This method represents the first application of α-amino boron reagents as reaction partners in rhodium-catalyzed couplings. Reactions with trifluoromethyl ketones proceed in reasonable yields and with good diastereoselectivity along with complete retention at the organoboron stereocenter. The potential of this method is further highlighted by the exploration of a variety of nitrogen substituents and addition to benzaldehyde and trityl-protected isatin.
Co-reporter:Jeffrey A. Boerth and Jonathan A. Ellman
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:
Publication Date(Web):
DOI:10.1039/C5SC04138D



![4-({[(2-Methyl-2-propanyl)oxy]carbonyl}amino)-2-butynoic acid](http://img.cochemist.com/ccimg/168800/168762-94-5.png)
![4-({[(2-Methyl-2-propanyl)oxy]carbonyl}amino)-2-butynoic acid](http://img.cochemist.com/ccimg/168800/168762-94-5_b.png)

![Pyrrolidine, 1-[(1-methyl-1H-indol-3-yl)carbonyl]-](http://img.cochemist.com/ccimg/137700/137643-32-4.png)
![Pyrrolidine, 1-[(1-methyl-1H-indol-3-yl)carbonyl]-](http://img.cochemist.com/ccimg/137700/137643-32-4_b.png)


![Silane, [5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-pentynyl]trimethyl-](http://img.cochemist.com/ccimg/110600/110519-13-6.png)
![Silane, [5-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1-pentynyl]trimethyl-](http://img.cochemist.com/ccimg/110600/110519-13-6_b.png)
![2-Butenoic acid, 4-[[(1,1-dimethylethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/89800/89711-09-1.png)
![2-Butenoic acid, 4-[[(1,1-dimethylethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/89800/89711-09-1_b.png)







![Ethanone, 1-[1,1'-biphenyl]-3-yl-2-bromo-](/data/chemimg/3389900/73932-62-4.png)
![Ethanone, 1-[1,1'-biphenyl]-3-yl-2-bromo-](/data/chemimg/3389900/73932-62-4_b.png)


















![1-methyl-3-[(Z)-2-nitroethenyl]-1H-indole](http://img.cochemist.com/ccimg/2800/2731-00-2.png)
![1-methyl-3-[(Z)-2-nitroethenyl]-1H-indole](http://img.cochemist.com/ccimg/2800/2731-00-2_b.png)




![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)


















![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7.png)
![N-[3,5-Bis(trifluoromethyl)phenyl]-N-[(8a,9S)-6-methoxy-9-cinchonanyl]thiourea](/data/chemimg/103500/852913-16-7_b.png)




![Silane,trimethyl[[(2E)-2-methyl-3-phenyl-1-(trifluoromethyl)-2-propenyl]oxy]-](http://img.cochemist.com/ccimg/659800/659721-44-5.png)
![Silane,trimethyl[[(2E)-2-methyl-3-phenyl-1-(trifluoromethyl)-2-propenyl]oxy]-](http://img.cochemist.com/ccimg/659800/659721-44-5_b.png)


































![2-Butenoic acid, 4-[[(1,1-dimethylethoxy)carbonyl]amino]-, methyl ester](http://img.cochemist.com/ccimg/139300/139200-36-5.png)
![2-Butenoic acid, 4-[[(1,1-dimethylethoxy)carbonyl]amino]-, methyl ester](http://img.cochemist.com/ccimg/139300/139200-36-5_b.png)


![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)










![Benzaldehyde, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-95-5.png)
![Benzaldehyde, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-95-5_b.png)
![Benzenemethanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-77-3.png)
![Benzenemethanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-77-3_b.png)




















![4,5-dihydro-9,10-dimethoxypyrrolo[3,2,1-de]phenanthridin-7-one](http://img.cochemist.com/ccimg/65400/65367-74-0.png)
![4,5-dihydro-9,10-dimethoxypyrrolo[3,2,1-de]phenanthridin-7-one](http://img.cochemist.com/ccimg/65400/65367-74-0_b.png)










![N-[3-(2,5-dioxo-4-imidazolidinyl)propyl]-Guanidine](http://img.cochemist.com/ccimg/59000/58942-06-6.png)
![N-[3-(2,5-dioxo-4-imidazolidinyl)propyl]-Guanidine](http://img.cochemist.com/ccimg/59000/58942-06-6_b.png)




















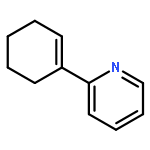
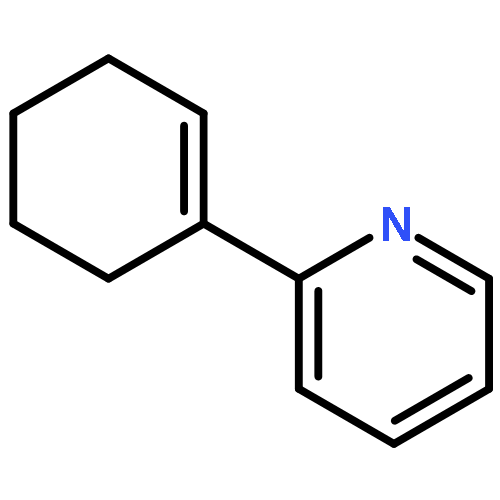






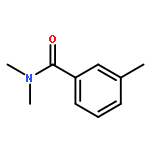
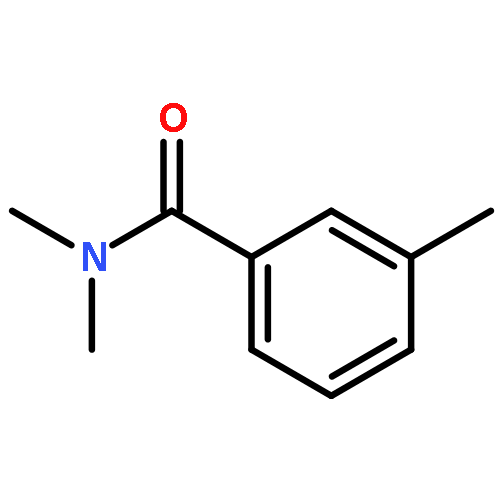






















![Ethanone,1-[1,1'-biphenyl]-3-yl-](http://img.cochemist.com/ccimg/3200/3112-01-4.png)
![Ethanone,1-[1,1'-biphenyl]-3-yl-](http://img.cochemist.com/ccimg/3200/3112-01-4_b.png)





























![2,6-BIS(METHYLSULFONYL)-2,6-DIAZASPIRO[3.3]HEPTANE](http://img.cochemist.com/ccimg/74800/74786-81-5.png)
![2,6-BIS(METHYLSULFONYL)-2,6-DIAZASPIRO[3.3]HEPTANE](http://img.cochemist.com/ccimg/74800/74786-81-5_b.png)
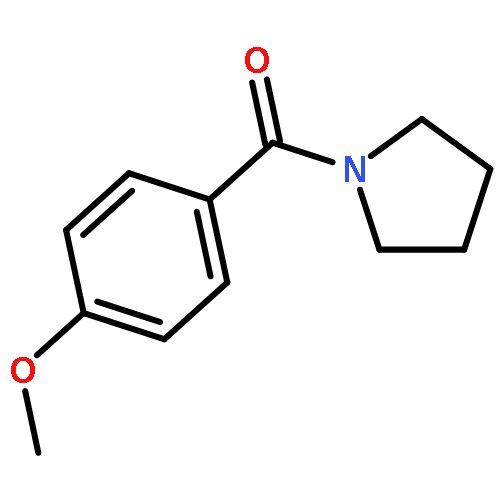








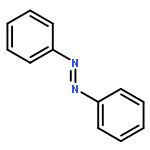
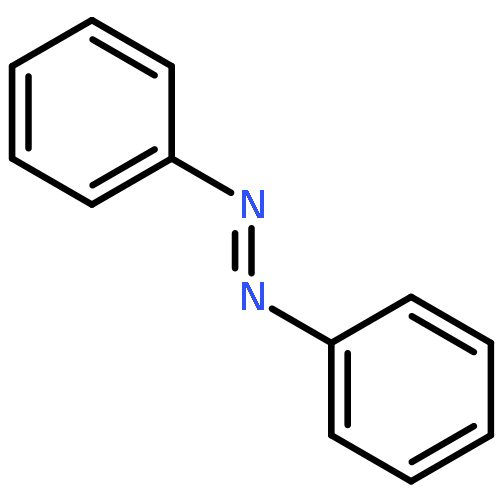




![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)