Co-reporter:Imtiaz Khan, Mattia Manzotti, Graham J. Tizzard, Simon J. Coles, Rebecca L. Melen, and Louis C. Morrill
ACS Catalysis November 3, 2017 Volume 7(Issue 11) pp:7748-7748
Publication Date(Web):October 16, 2017
DOI:10.1021/acscatal.7b03077
Herein we report the metal-free diastereoselective frustrated Lewis pair (FLP)-catalyzed hydrogenation of aza-Morita–Baylis–Hillman (aza-MBH) adducts, accessing a diverse range of stereodefined β-amino acid derivatives in excellent isolated yields (28 examples, 89% average yield, up to 90:10 d.r.). Furthermore, sequential organo-FLP catalysis has been developed. An initial organocatalyzed aza-MBH reaction followed by in situ FLP formation and hydrogenation of the electron-deficient α,β-unsaturated carbonyl compounds can be performed in one-pot, using DABCO as the Lewis base in both catalytic steps.Keywords: amino esters; frustrated Lewis pairs; metal-free hydrogenation; sequential catalysis; stereoselective;
Co-reporter:Qin Yin, Yashar Soltani, Rebecca L. Melen, and Martin Oestreich
Organometallics July 10, 2017 Volume 36(Issue 13) pp:2381-2381
Publication Date(Web):June 19, 2017
DOI:10.1021/acs.organomet.7b00381
The rarely used boron Lewis acid tris[3,5-bis(trifluoromethyl)phenyl]borane (BArF3) is found to be an excellent catalyst for metal-free hydroboration of imines. In the presence of 1.0 mol % of BArF3, several ketimines and aldimines undergo hydroboration with pinacolborane (HBpin) at room temperature without the aid of an external Lewis base. BArF3 is more reactive than other Lewis acidic boranes, including the often-used tris(pentafluorophenyl)borane (B(C6F5)3). The steric hindrance imparted by the six fluorine atoms ortho to the boron center in B(C6F5)3 accounts for this. Mechanistic control experiments indicate conventional Lewis acid catalysis involving imine activation and hydride transfer from HBpin.
Co-reporter:James R. Lawson and Rebecca L. Melen
Inorganic Chemistry August 7, 2017 Volume 56(Issue 15) pp:8627-8627
Publication Date(Web):February 3, 2017
DOI:10.1021/acs.inorgchem.6b02911
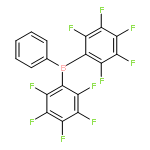
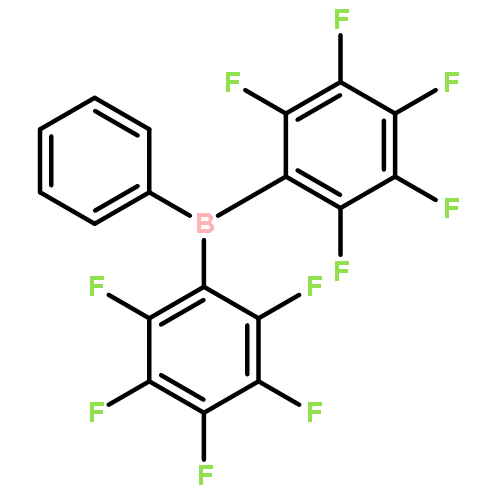
As main-group chemistry, in particular boron chemistry, has expanded and developed over the past 20 years, one reagent has risen to prominence as well. Tris(pentafluorophenyl)borane, B(C6F5)3 (commonly known as BCF), has demonstrated extensive applications in a wide variety of reactions, including borylation, hydrogenation, hydrosilylation, frustrated Lewis pair (FLP) chemistry, Lewis acid catalysis, and more. The high Lewis acidity of B(C6F5)3 is derived from the electronic effects of its three C6F5 rings, rendering it a versatile reagent for a great number of reactions. In addition, the steric bulk of these rings also allows it to function as the Lewis acid in a FLP, granting this reagent yet another synthetically useful application. However, as main-group chemistry continues to evolve as a field, new reagents are required that go beyond BCF, increasing not only the range of reactions available but also the breadth of compounds attainable. Great strides have already been made in order to accomplish this task, and this review will highlight modern advances in boron chemistry relating to borylation reactions. Herein, we will show the recent uses of B(C6F5)3 in borylation reactions while also focusing on current advances in novel borane and borocation usage that eclipses that of the stalwart B(C6F5)3.
Co-reporter:Yashar Soltani;Lewis C. Wilkins;Dr. Rebecca L. Melen
Angewandte Chemie 2017 Volume 129(Issue 39) pp:12157-12161
Publication Date(Web):2017/09/18
DOI:10.1002/ange.201704789
AbstractDiese Arbeit beschreibt eine neuartige katalytische Cyclisierung unter Verwendung eines stark Lewis-sauren Borans mit einhergehender C-H- oder C-C-Bindungsbildung. Die Aktivierung von Molekülen mit C-C-Dreifachbindungen unter Verwendung von B(C6F5)3 ermöglicht erstmals eine entsprechende Lewis-Säure-katalysierte, intramolekulare Cyclisierung von Carbonsäuren. Darüber hinaus zeigen die Ester dieser Carbonsäuren mit katalytischen Mengen B(C6F5)3 eine formale 1,5-Migration der Estergruppen unter Knüpfung einer C-C-Bindung. Unter Verwendung dieser neuen metallfreien Methode konnten anspruchsvolle Dihydropyrone und Isocumarine in hohen Ausbeuten unter milden Bedingungen sowie mit hoher Atomökonomie synthetisiert werden.
Co-reporter:Yashar Soltani;Lewis C. Wilkins;Dr. Rebecca L. Melen
Angewandte Chemie International Edition 2017 Volume 56(Issue 39) pp:11995-11999
Publication Date(Web):2017/09/18
DOI:10.1002/anie.201704789
AbstractThis work showcases a new catalytic cyclization reaction using a highly Lewis acidic borane with concomitant C−H or C−C bond formation. The activation of alkyne-containing substrates with B(C6F5)3 enabled the first catalytic intramolecular cyclizations of carboxylic acid substrates using this Lewis acid. In addition, intramolecular cyclizations of esters enable C−C bond formation as catalytic B(C6F5)3 can be used to effect formal 1,5-alkyl migrations from the ester functional groups to unsaturated carbon–carbon frameworks. This metal-free method was used for the catalytic formation of complex dihydropyrones and isocoumarins in very good yields under relatively mild conditions with excellent atom efficiency.
Co-reporter:Lewis C. Wilkins, Rebecca L. Melen
Coordination Chemistry Reviews 2016 Volume 324() pp:123-139
Publication Date(Web):1 October 2016
DOI:10.1016/j.ccr.2016.07.011
•An overview of emerging main group catalysts used in chiral induction is outlined.•Mechanistic aspects of various enantioselective catalytic cycles are described.•Solutions to common limitations of main group systems are offered.•Main group alternatives to traditional transition metal complexes are presented.This review highlights a number of recent developments in the field of main group enantioselective catalysis. Many essential transformations can be effected catalytically such as hydrosilylation, hydroamination and hydrogenation reactions, amongst others, in an asymmetric fashion using earth abundant s- and p-block elements such as calcium, strontium, boron and aluminum. Recent work in this area has shown that these systems are not only active in catalysis but may also have the potential to compete with transition metal based systems with the reduced cost and toxicity sometimes associated with main group chemistry.The current field of synthetic enantioselective catalysis is dominated by transition metals such as ruthenium, palladium and copper, launched through the success of pioneering work by Knowles, Noyori and Sharpless, however, there are inherent problems with using heavy rare earth transition metals, namely their prohibitive costs and potential toxicity. To combat this an alternative has been found in the form of main group centered catalysts. Indeed the recent application of main group elements in catalysis has expanded to encompass enantioselective transformations. As main group elements, for example calcium, magnesium, boron and aluminum have become more preeminent as active catalysts, their applications in enantioselective catalysis has also made great strides in recent years. In this review we highlight the importance of earth abundant main group s- and p-block catalysts as candidates for effective asymmetric induction in a range of organic transformations such as 1,4-additions, hydrosilylation, hydrogenation and phosphonylation as well as dispelling common myths surrounding early s- and p-block elements with regard to their catalytic applications.
Co-reporter:Rebecca L. Melen, Robert J. Less, Christopher M. Pask, and Jeremy M. Rawson
Inorganic Chemistry 2016 Volume 55(Issue 22) pp:11747-11759
Publication Date(Web):November 1, 2016
DOI:10.1021/acs.inorgchem.6b01771
The synthesis and structural characterization of a series of perfluoroaryldiselenadiazolyls [DSeDA; p-XC6F4CNSeSeN (X = F, Cl, Br, CF3, NO2, and CN for 2a–2f, respectively)] are described. Concentration-dependent solution UV/vis measurements on 2a follow the Beer–Lambert law and the transitions assigned through time-dependent density functional theory (TD-DFT) studies, indicating little propensity for dimerization in solution (10–3–10–4 M). Solution electron paramagnetic resonance (EPR) spectra reveal that these radicals exhibit a broad featureless singlet around g = 2.04 but form well-resolved anisotropic EPR spectra in frozen solution, from which spin densities were determined and found to reflect an increase in the spin density at the chalcogen in relation to the corresponding dithiadiazolyl (DTDA) radicals, p-XC6F4CNSSN. The solid-state structures of 2a and 2d–2f all adopt spin-paired cis-cofacial dimers in which the dimers are held together via multicenter π*−π* “pancake bonding” interactions. Conversely, 2b and 2c exhibit an orthogonal mode of association, which is unique to DSeDA chemistry but which also affords a singlet ground state evidenced by SQUID magnetometry. The more sterically demanding diselenadiazolyl radical 2f was also prepared and exhibits a trans-antarafacial dimerization mode. DFT studies [UPBE0-D3 ccPVTZ-PP(-F)++] on the model radical HCNSeSeN confirm that each dimer is a stable energy minimum on the potential energy surface, reproducing well the experimental geometric parameters with relative stability in the order cis-cofacial > orthogonal > trans-antarafacial. Computational studies reflect stronger dimerization for DSeDA radicals in relation to their sulfur analogues, consistent with the experimental observation: While 2a and 2d are isomorphous with their corresponding DTDA radicals, 2b, 2c, and 2e–2g are all dimeric, in contrast to their DTDA analogues, which are monomeric in the solid-state. A study on 2f reveals that significant geometric strain accumulates in order to support the propensity for both cis dimerization and intermolecular CN···Se interactions. Conversely, p-NCC6F4CNSSN likely forfeits dimerization in the analogous packing motif in order to release strain but retains the favorable intermolecular CN···S interactions.
Co-reporter:Lewis C. Wilkins, Hugh B. Hamilton, Benson M. Kariuki, A. Stephen K. Hashmi, Max M. Hansmann and Rebecca L. Melen
Dalton Transactions 2016 vol. 45(Issue 14) pp:5929-5932
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5DT03340C
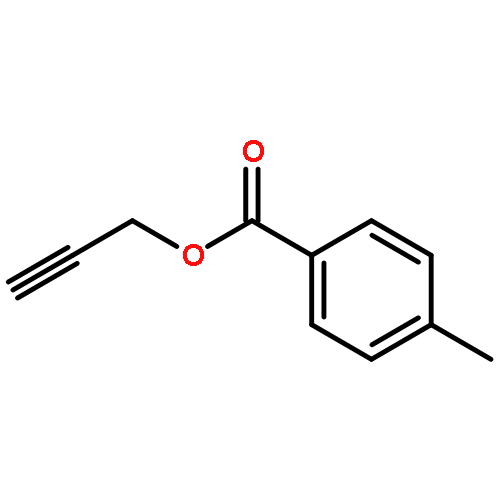
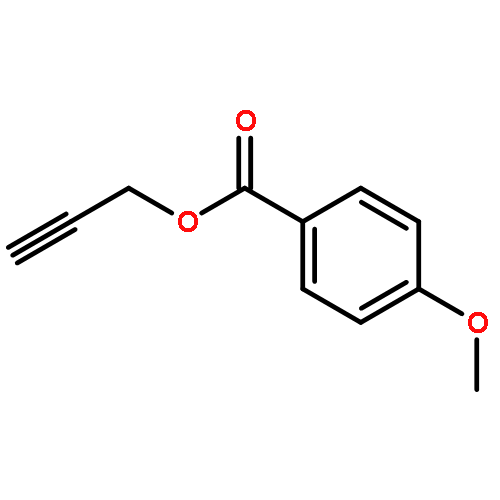
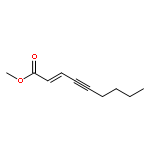
Treatment of methyl (Z)-2-alken-4-ynoates with the strong Lewis acid tris(pentafluorophenyl) borane, B(C6F5)3, yield substituted zwitterionic pyrylium borate species via an intramolecular 6-endo-dig cyclisation reaction.
Co-reporter:Rebecca L. Melen, Lewis C. Wilkins, Benson M. Kariuki, Hubert Wadepohl, Lutz H. Gade, A. Stephen K. Hashmi, Douglas W. Stephan, and Max M. Hansmann
Organometallics 2015 Volume 34(Issue 16) pp:4127-4137
Publication Date(Web):August 13, 2015
DOI:10.1021/acs.organomet.5b00546
The reactions of allenes with frustrated (or cooperative) Lewis acid/base pairs result in the 1,4-addition of the base pair to the allene. The reactions of allenyl ketones and esters just in the presence of the strong Lewis acid B(C6F5)3 afford the selective formation of the 1,2-carboboration products. In both cases the Lewis acid activates the allene to either a C6F5 migration or nucleophilic attack by the Lewis base. In addition to the 1,2-carboboration pathway, which can be viewed as being triggered by activation of the ketone (σ-activation), in the case of allenyl esters the corresponding cyclization products are observed in the presence of water.
Co-reporter:Lewis C. Wilkins, Philipp Wieneke, Paul D. Newman, Benson M. Kariuki, Frank Rominger, A. Stephen K. Hashmi, Max M. Hansmann, and Rebecca L. Melen
Organometallics 2015 Volume 34(Issue 21) pp:5298-5309
Publication Date(Web):October 21, 2015
DOI:10.1021/acs.organomet.5b00753
The reactions of propargyl amides, ureas, carbamates, and carbonates with B(C6F5)3 proceed via an intramolecular 5-exo-dig cyclization across the alkyne unit to yield the corresponding vinyl borate species. The generated sp2 carbocation is stabilized by the flanking heteroatoms, allowing for isolation of oxazoline intermediates. The fate of these intermediates is strongly dependent upon the propargyl-functionalized starting material, with the carbamates and carbonates undergoing a ring-opening mechanism (propargyl rearrangement) to give cyclic allylboron compounds, while prolonged heating of the urea derivatives shows evidence of oxazole formation. In a deviation away from the reactivity of carbamates stated previously, the benzyl carbamate substrate undergoes dealkylation at the benzylic position, liberating 5-methyloxazol-2-(3H)-one.
Co-reporter:James R. Lawson, Lewis C. Wilkins, Manon André, Emma C. Richards, Mohammed N. Ali, James A. Platts and Rebecca L. Melen
Dalton Transactions 2016 - vol. 45(Issue 41) pp:NaN16181-16181
Publication Date(Web):2016/09/28
DOI:10.1039/C6DT03360A
A series of borocations have been synthesised from the addition of haloboranes to synthetically accessible N,N′-1,4-diazabutadiene precursors, which are derived from commercially available anilines. The synthesis and structural studies of the borocations are described.
Co-reporter:Lewis C. Wilkins, Hugh B. Hamilton, Benson M. Kariuki, A. Stephen K. Hashmi, Max M. Hansmann and Rebecca L. Melen
Dalton Transactions 2016 - vol. 45(Issue 14) pp:NaN5932-5932
Publication Date(Web):2015/09/25
DOI:10.1039/C5DT03340C
Treatment of methyl (Z)-2-alken-4-ynoates with the strong Lewis acid tris(pentafluorophenyl) borane, B(C6F5)3, yield substituted zwitterionic pyrylium borate species via an intramolecular 6-endo-dig cyclisation reaction.
Co-reporter:Jolie Lam, Benjamin A. R. Günther, Jeffrey M. Farrell, Patrick Eisenberger, Brian P. Bestvater, Paul D. Newman, Rebecca L. Melen, Cathleen M. Crudden and Douglas W. Stephan
Dalton Transactions 2016 - vol. 45(Issue 39) pp:NaN15316-15316
Publication Date(Web):2016/06/23
DOI:10.1039/C6DT02202B
The carbene derived from (1R,3S)-camphoric acid was used to prepare the borane adduct with Piers’ borane 7. Subsequent hydride abstraction gave the borenium cation 8. Adducts with 9-BBN and the corresponding (1R,3S)-camphoric acid-derived carbene bearing increasingly sterically demanding N-substituents (R = Me 9, Et 10, i-Pr 11) and the corresponding borenium cations 12–14 were also prepared. These cations were not active as catalysts in hydrogenation, although 9–11 were shown to undergo carbene ring expansion reactions at 50 °C to give species 15–17. The IBOX-carbene precursors 18 and 19 derived from amino alcohols (S)-valinol and (S)-tert-leucinol (R = i-Pr, t-Bu) were used to prepare borane adducts 20–23. Reaction of the carbenes 1,3-dimethylimidazol-2-ylidene (IMe), 1,3-di-iso-propylimidazol-2-ylidene (IPr) 1-benzyl-3-methylimidazol-2-ylidene (IBnMe), 1-methyl-3-phenylimidazol-2-ylidene (IPhMe) and 1-tert-butyl-3-methylimidazol-2-ylidene (ItBuMe) with diisopinocampheylborane (Ipc2BH) gave chiral adducts: (IMe)(Ipc2BH) 24, (IPr)(Ipc2BH) 25, (IBnMe)(Ipc2BH) 26, (IPhMe)(Ipc2BH) 27, and (ItBuMe)(Ipc2BH) 28. Triazolylidene-type adducts including the (10)-phenyl-9-borabicyclo [3.3.2]decane adduct of 1,3,4-triphenyl-1H-1,2,3-triazolium, rac-29 and the 9-BBN derivative of (S)-2-amino-2′-methoxy-1,1′-binaphthalene-1,2,3-triazolium 34a/b were also prepared. In catalytic studies of these systems, while several species were competent catalysts for imine reduction, in general, low enantioselectivities, ranging from 1–20% ee, were obtained. The implications for chiral borenium cation catalyst design are considered.















![Quinoline, 8-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/23200/23111-13-9.png)
![Quinoline, 8-[(trimethylsilyl)oxy]-](http://img.cochemist.com/ccimg/23200/23111-13-9_b.png)