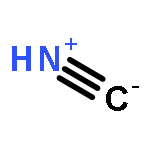
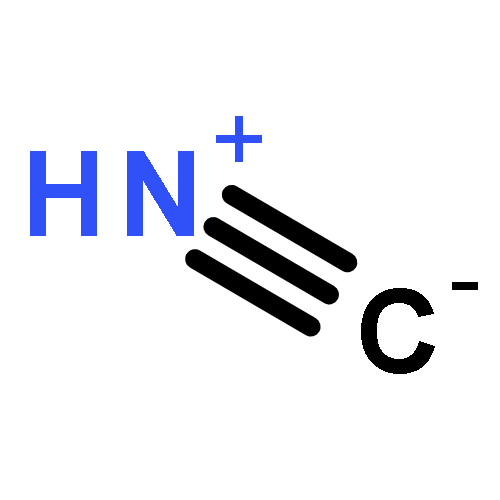
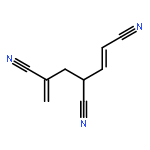
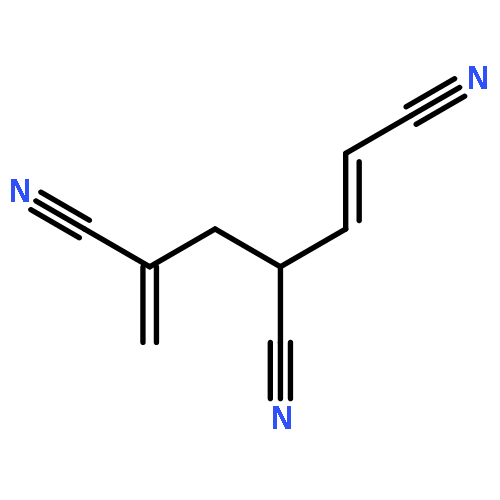
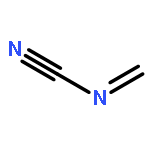
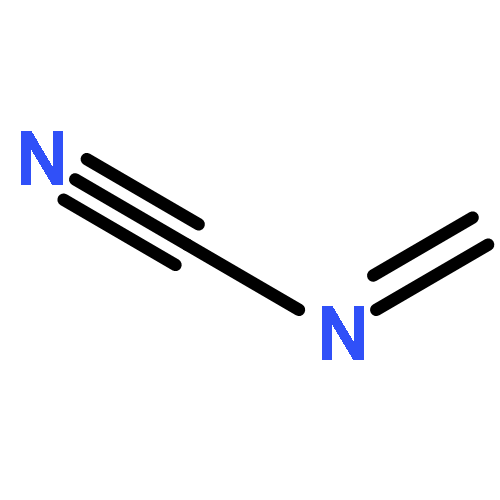
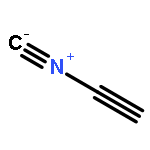
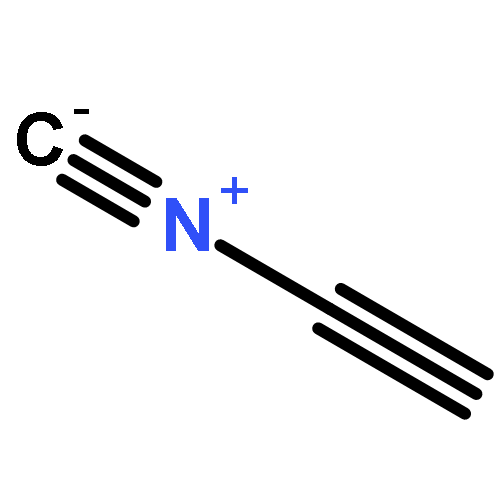
Co-reporter:Peter B. Karadakov, David L. Cooper
Computational and Theoretical Chemistry 2017 Volume 1116(Volume 1116) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.comptc.2017.01.003
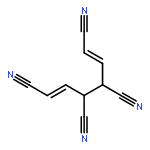
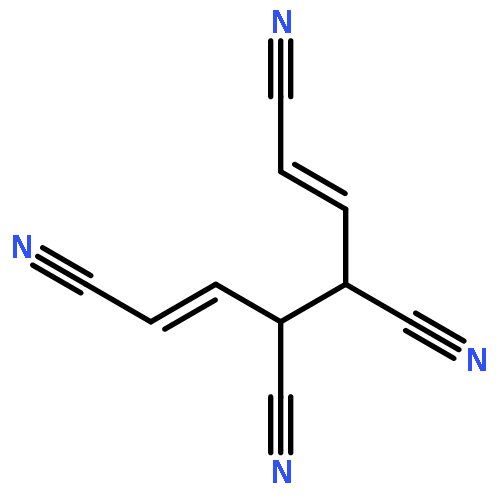
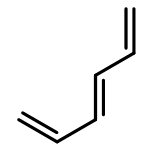
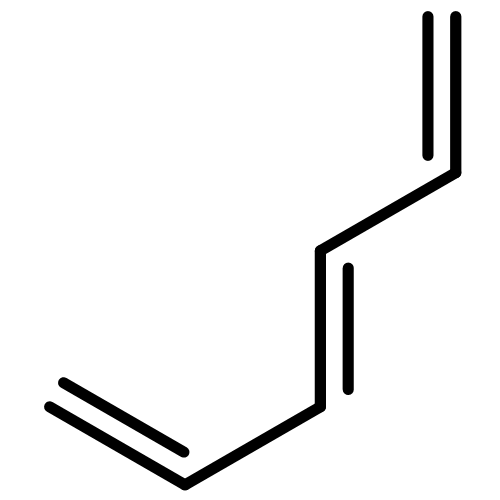
•Five 10-π-electron fused conjugated systems with cyclopropenyl rings are planar (MP2).•Spin-coupled calculations provide familiar patterns of well-localized active orbitals.•Aromatic ground states: in-phase combination of two “’Kekulé” Rumer structures.•First singlet electronic excited states probably antiaromatic: “antiresonance”.The feasibilities and electronic structures of five ten-π-electron fused conjugated molecules involving cyclopropenyl rings are explored using second-order Møller-Plesset perturbation theory (MP2), spin-coupled (SC) and complete-active-space self-consistent-field (CASSCF) wavefunctions, in the cc-pVTZ basis. All five fused conjugated molecules are predicted to have rigid planar ground state geometries of C2v or D2h symmetry and large dipole moments (if not of D2h symmetry). The compact ground state SC(10) wavefunctions with ten active orbitals for these molecules are found to be of comparable quality to the respective CASSCF(10,10) constructions, but much easier to interpret. The analyses of the ground state SC(10) wavefunctions for all five fused conjugated molecules reveal resonance patterns which indicate that all of these molecules are aromatic in their electronic ground states; on the other hand, the SC(10) approximations to the first singlet electronic excited states are found to exhibit “antiresonance” which suggests that each of the five molecules switches from aromatic to antiaromatic upon vertical excitation from the ground state to its first singlet excited state. Ring strain prevents the formation of a fused structure involving three cyclopropenyl rings and a cycloheptatrienyl ring; the alternative stable dehydro compound which resembles m-benzyne is shown, using a SC(12) wavefunction, to involve a weak σ bond between the dehydro centres.Download high-res image (206KB)Download full-size image
Co-reporter:Peter B. Karadakov and Kate E. Horner
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 2) pp:558-563
Publication Date(Web):January 6, 2016
DOI:10.1021/acs.jctc.5b00842
Differences in nuclear isotropic magnetic shieldings give rise to the chemical shifts measured in NMR experiments. In contrast to existing NMR experimental techniques, quantum chemical methods are capable of calculating isotropic magnetic shieldings not just at nuclei, but also at any point in the space surrounding a molecule. Using s-trans-1,3-butadiene, ethane, ethene, and ethyne as examples, we show that the variations in isotropic magnetic shielding around a molecule, represented as isosurfaces and contour plots, provide an unexpectedly clear picture of chemical bonding, which is much more detailed than the traditional description in terms of the total electron density.
Co-reporter:Peter B. Karadakov
Chemical Physics Letters 2016 Volume 646() pp:190-196
Publication Date(Web):16 February 2016
DOI:10.1016/j.cplett.2015.12.068
Highlights
- •
Vibrational frequencies of C6n2H6n (n = 2–12) coronenes have been calculated.
- •
C6n2H6n coronenes are expected to switch to a nonplanar geometry at around n = 9–12.
- •
A single graphene sheet is predicted to bend by sagging in the middle.
- •
Popular methods (Hartree–Fock and DFT) can produce anomalous imaginary frequencies.
- •
Calculations on extended systems require further basis set development.
Co-reporter:Peter B. Karadakov and David L. Cooper
The Journal of Physical Chemistry A 2016 Volume 120(Issue 43) pp:8769-8779
Publication Date(Web):October 14, 2016
DOI:10.1021/acs.jpca.6b09426
Spin-coupled (SC) theory is used to obtain modern valence-bond descriptions of the electronic structures of local minimum and transition-state geometries of three species that have been considered to exhibit homoconjugation and homoaromaticity: the homotropenylium ion, C8H9+, the cycloheptatriene neutral ring, C7H8, and the 1,3-bishomotropenylium ion, C9H11+. The resulting compact SC wave functions are of comparable quality to complete-active-space self-consistent field constructions that are based on the same “N electrons in M orbitals” active spaces, but they are much easier to interpret directly. Analysis of the forms of the SC orbitals and of the overlaps between them, as well as an examination of the compositions of the associated resonance patterns, strongly suggest that both of the homotropenylium and 1,3-bishomotropenylium ions are homoaromatic at their local minimum geometries, with all of the other cases that were considered being nonaromatic. The SC results also show that the differences between “no-bond” and “bond” homoconjugated systems are very likely to be much smaller than previously thought.
Co-reporter:Peter B. Karadakov, Peter Hearnshaw, and Kate E. Horner
The Journal of Organic Chemistry 2016 Volume 81(Issue 22) pp:11346-11352
Publication Date(Web):October 27, 2016
DOI:10.1021/acs.joc.6b02460
Aromaticity, antiaromaticity, and their effects on chemical bonding in the ground states (S0), lowest triplet states (T1), and the first and second singlet excited states (S1 and S2) of benzene (C6H6) and square cyclobutadiene (C4H4) are investigated by analyzing the variations in isotropic magnetic shielding around these molecules in each electronic state. All shieldings are calculated using state-optimized π-space complete-active-space self-consistent field (CASSCF) wave functions constructed from gauge-including atomic orbitals (GIAOs), in the 6-311++G(2d,2p) basis. It is shown that the profoundly different shielding distributions in the S0 states of C6H6 and C4H4 represent aromaticity and antiaromaticity “fingerprints” which are reproduced in other electronic states of the two molecules and allow classification of these states as aromatic (S0 and S2 for C6H6, T1 and S1 for C4H4) or antiaromatic (S0 and S2 for C4H4, T1 and S1 for C6H6). S2 C6H6 is predicted to be even more aromatic than S0 C6H6. As isotropic shielding isosurfaces and contour plots show very clearly the effects of aromaticity and antiaromaticity on chemical bonding, these can be viewed, arguably, as the most succinct visual definitions of the two phenomena currently available.
Co-reporter:Peter B. Karadakov
Applied Organometallic Chemistry 2015 Volume 29( Issue 1) pp:
Publication Date(Web):
DOI:10.1002/aoc.3239
Co-reporter:Kate E. Horner and Peter B. Karadakov
The Journal of Organic Chemistry 2015 Volume 80(Issue 14) pp:7150-7157
Publication Date(Web):June 17, 2015
DOI:10.1021/acs.joc.5b01010
Isotropic magnetic shielding distributions in the regions of space surrounding oxazole, imidazole, and thiazole are used to investigate aromaticity and bonding in these five-membered heterocycles with two heteroatoms. This is achieved by constructing HF-GIAO and MP2-GIAO (Hartree–Fock and second-order Møller–Plesset perturbation theory with gauge-including atomic orbitals) isotropic shielding plots, within the 6-311++G(d,p) basis, using regular two-dimensional 0.05 Å grids in the molecular plane and in planes 0.5 and 1 Å above it. The extent of isotropic shielding delocalization in the contour plots in planes 1 Å above the molecular plane, which is a new sensitive two-dimensional aromaticity criterion, indicates that aromaticity decreases in the order thiazole > imidazole > oxazole; in combination with previous results on furan, pyrrole, and thiophene ( J. Org. Chem. 2013, 78, 8037−9043), the aromaticity ordering in the six five-membered heterocycles becomes thiophene > thiazole > pyrrole > imidazole > furan > oxazole. The results suggest that the inclusion of a second heteroatom in a five-membered heterocycle has a detrimental effect on its aromaticity, which is very minor in oxazole, when compared to furan, and small but noticeable in imidazole and pyrrole and in thiazole and thiophene.
Co-reporter:Linda J. M
c
Allister, Duncan W. Bruce and Peter B. Karadakov
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 6) pp:2576-2587
Publication Date(Web):02 Dec 2013
DOI:10.1039/C3CP54612H
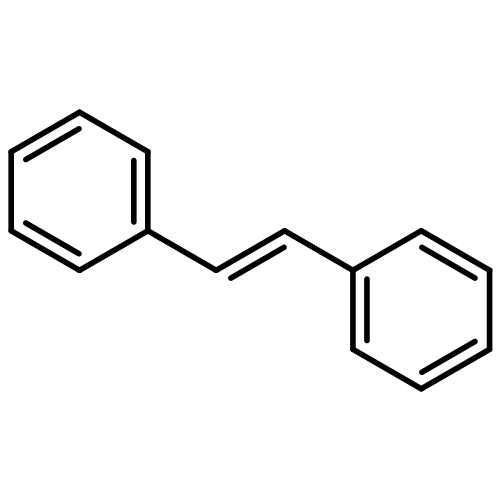
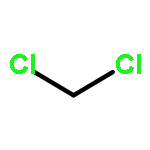
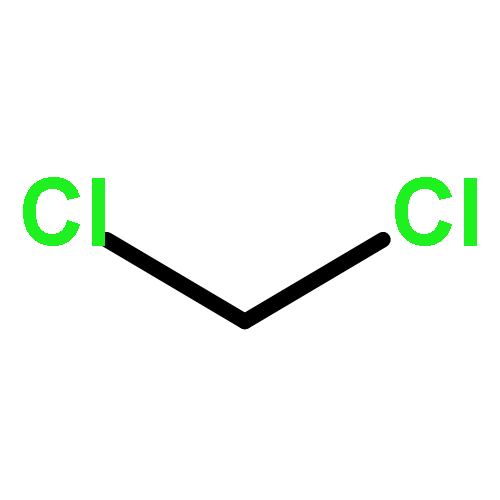
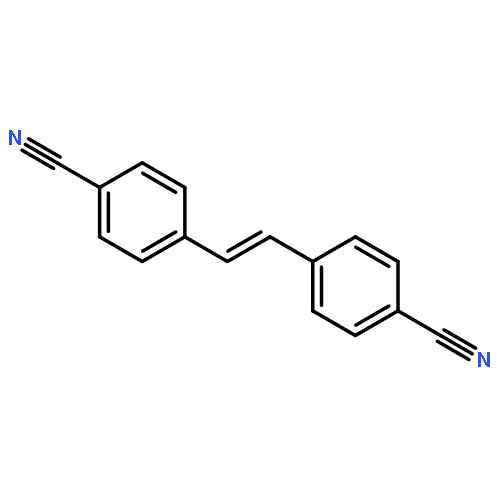
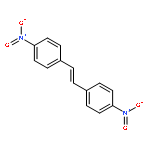
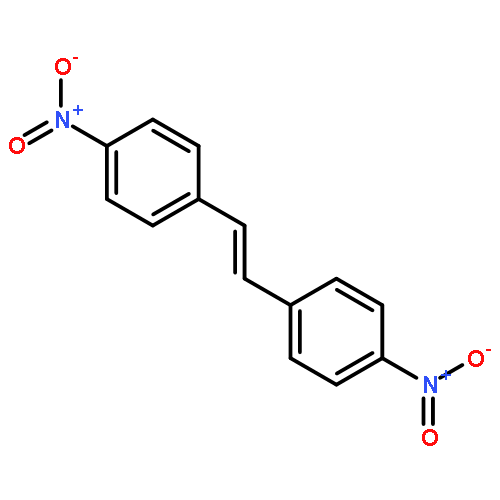
In order to study the effect of substituents on the electrophilic bromination of stilbenes and stilbazoles, the geometries of the reaction intermediates were optimised at the MP2/6-31G(d,p) and M06-2X/6-31G(d,p) levels of theory. Stilbenes with two electron-withdrawing substituents are shown to favour bromonium ion intermediates, whereas the presence of an electron-donating substituent leads to a carbocation intermediate. These observations are rationalised by means of Hammett and Taft parameters. Localised molecular orbitals indicate that the bonding between the bromine atom and the carbon atoms in the stilbene varies from a σ-bond in a carbocation intermediate to a 3-centre-2-electron bond in a bromonium ion intermediate. Natural bond orbital (NBO) analyses reveal that carbocation intermediates are stabilised by resonance. The optimised geometries of charge-transfer complexes, the pre-reactive intermediates formed between molecular bromine and the stilbene, show that these species can be considered as halogen-bonded complexes with binding energies ranging from 14.5 to 20.1 kJ mol−1.
Co-reporter:Peter B. Karadakov and Kate E. Horner
The Journal of Physical Chemistry A 2013 Volume 117(Issue 2) pp:518-523
Publication Date(Web):December 17, 2012
DOI:10.1021/jp311536c
A detailed picture of the variations in the isotropic shielding σiso(r) in and around the classical examples of aromatic and antiaromatic systems, benzene and square cyclobutadiene, in terms of isosurfaces and contour plots, is obtained by calculating σiso(r) values at fine regular three-dimensional 7 × 7 × 7 Å grids of points with a spacing of 0.05 Å, using π space complete-active-space self-consistent field (CASSCF) wave functions constructed from gauge-including atomic orbitals (GIAOs). The results demonstrate that the σiso(r) values can be used not only to distinguish between aromatic and antiaromatic systems but also to characterize chemical bonds and investigate the extents to which these bonds are affected by the aromatic or antiaromatic nature of the molecule in which they reside. The strong bonding interactions within the benzene ring are highlighted by the fact that the carbon–carbon and carbon–hydrogen bonds are wrapped up within a doughnut-shaped region of increased shielding, which is especially high along the carbon–carbon bonds, with protrusions marking the carbon–hydrogen bonds. The antiaromatic destabilization in square cyclobutadiene is seen as the consequence of the presence of a markedly deshielded dumbbell-shaped region in the center of the molecule, which disrupts the connections between the shielded regions outlining individual carbon–carbon bonds, decreases the shielding within these regions, and displaces them to off-bond locations outside the ring. The well-known deshielding of aromatic protons and increased shielding of antiaromatic protons are shown to be accompanied by analogous shielding differences along the whole lengths of the carbon–hydrogen bonds, as a result of which the carbon–hydrogen bonds should become slightly weaker in aromatic systems as opposed to the corresponding bonds in antiaromatic systems.
Co-reporter:Kate E. Horner and Peter B. Karadakov
The Journal of Organic Chemistry 2013 Volume 78(Issue 16) pp:8037-8043
Publication Date(Web):July 23, 2013
DOI:10.1021/jo401319k
Aromaticity and bonding in furan, pyrrole, and thiophene are investigated through the behavior of the isotropic shielding σiso(r) within the regions of space surrounding these molecules. HF-GIAO/6-311++G(d,p) and MP2-GIAO/6-311++G(d,p) (Hartree–Fock and second-order Møller–Plesset perturbation theory utilizing gauge-including atomic orbitals) σiso(r) contour plots are constructed using regular two-dimensional 0.05 Å grids in the molecular plane, in horizontal planes 0.5 and 1 Å above it, and in a vertical plane through the heteroatom. The nucleus-independent chemical shifts (NICS) calculated at the ring centers and at 0.5 Å and 1 Å above these centers, NICS(0), NICS(0.5), and NICS(1), respectively, support the widely accepted order of aromaticities thiophene > pyrrole > furan. The results suggest that accurate NICS calculations benefit more from the use of an extended basis set than from the inclusion of dynamical electron correlation effects. The different extents of σiso(r) delocalization observed in the horizontal contour plots and other features of σiso(r) are also consistent with an aromaticity reduction of the order thiophene > pyrrole > furan. It is suggested that the extent of σiso(r) delocalization in σiso(r) contour plots in planes 1 Å above the molecular plane could be used for comparing the relative aromaticities of a wide range of aromatic systems.
Co-reporter:Peter B. Karadakov, David L. Cooper, Brian J. Duke, and Jiabo Li
The Journal of Physical Chemistry A 2012 Volume 116(Issue 26) pp:7238-7244
Publication Date(Web):June 12, 2012
DOI:10.1021/jp303998h
Spin-coupled (SC) theory, an ab initio valence bond (VB) approach which uses a compact and an easy-to-interpret single-orbital product wave function comparable in quality to a ‘N in N’ complete-active-space self-consistent field [CASSCF(N,N)] construction, is extended to ‘N in M’ (N ≠ M) active spaces. The SC(N,M) wave function retains the essential features of the original SC model: It involves just the products of nonorthogonal orbitals covering all distributions of N electrons between M orbitals in which as few orbitals as possible, |N – M|, are doubly occupied (for N > M) or missing (for N < M) and all other orbitals are singly occupied; each of these products is combined with a flexible spin function which allows any mode of coupling of the spins of the orbitals within the product. The SC(N,M) wave function remains much more compact than a CASSCF(N,M) construction; for example, the SC(6,7) wave function includes 35 configuration state functions (CSFs) as opposed to the 490 CSFs in the CASSCF case. The essential features of the SC(N,M) method are illustrated through a SC(6,5) calculation on the cyclopentadienyl anion, C5H5–, and a SC(6,7) calculation on the tropylium cation, C7H7+. The SC(6,5) and SC(6,7) wave functions for C5H5– and C7H7+ are shown to provide remarkably clear modern VB models for the electronic structures of these aromatic cyclic ions which closely resemble the well-known SC model of benzene and yet recover almost all of the correlation energy included in the corresponding CASSCF(6,5) and CASSCF(6,7) wave functions: over 97% in the case of C5H5– and over 95% in the case of C7H7+.
Co-reporter:Linda J. McAllister, Duncan W. Bruce, and Peter B. Karadakov
The Journal of Physical Chemistry A 2011 Volume 115(Issue 40) pp:11079-11086
Publication Date(Web):September 2, 2011
DOI:10.1021/jp207119c
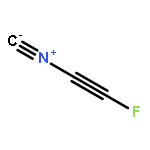
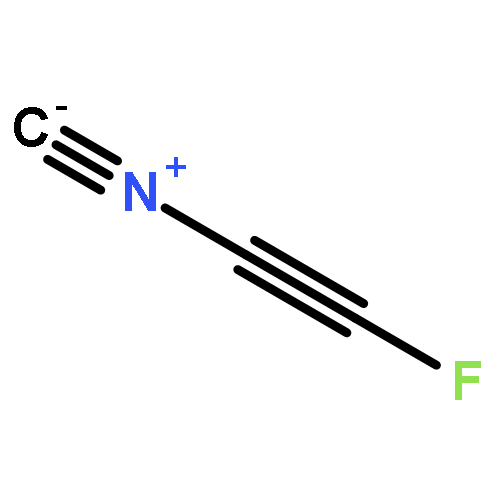
The optimized geometries and corresponding binding energies of complexes between fluorohalides, FX (X = Cl, Br, and I), and isocyanides, CNY (Y = CN, NC, NO2, F, CF3, Cl, Br, H, CCF, CCH, CH3, SiH3, Li, and Na), were calculated at the MP2(Full)/aug-cc-pVTZ (aug-cc-pVTZ-PP on I) level of theory, without and with basis set superposition error (BSSE) corrections through the counterpoise (CP) method. The optimized complex geometries were analyzed through the Steiner–Limbach relationship, which can be used to establish correlations between the F–X and X–C bond lengths. For all complexes, the correlations were shown to improve considerably when using optimized geometries including BSSE corrections. It was shown that further improvements can be achieved through the introduction of an extended four-parameter form of the Steiner–Limbach relationship which accounts for all differences between the valences associated with the two bonds involving the halogen in an A–X···B complex. The results indicate that chlorine as a halogen bond donor is affected by the basicity of the isocyanides and forms different types of halogen bonds as the F–Cl bond lengthens in parallel with the shortening of the distance between Cl and the isocyanide carbon. This is not observed for iodine and bromine as halogen-bond donors, which is illustrated by the low levels of correlation obtained when applying the standard and extended Steiner–Limbach relationships to the corresponding complexes.
Co-reporter:Linda J. M
c
Allister, Duncan W. Bruce and Peter B. Karadakov
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 6) pp:NaN2587-2587
Publication Date(Web):2013/12/02
DOI:10.1039/C3CP54612H
In order to study the effect of substituents on the electrophilic bromination of stilbenes and stilbazoles, the geometries of the reaction intermediates were optimised at the MP2/6-31G(d,p) and M06-2X/6-31G(d,p) levels of theory. Stilbenes with two electron-withdrawing substituents are shown to favour bromonium ion intermediates, whereas the presence of an electron-donating substituent leads to a carbocation intermediate. These observations are rationalised by means of Hammett and Taft parameters. Localised molecular orbitals indicate that the bonding between the bromine atom and the carbon atoms in the stilbene varies from a σ-bond in a carbocation intermediate to a 3-centre-2-electron bond in a bromonium ion intermediate. Natural bond orbital (NBO) analyses reveal that carbocation intermediates are stabilised by resonance. The optimised geometries of charge-transfer complexes, the pre-reactive intermediates formed between molecular bromine and the stilbene, show that these species can be considered as halogen-bonded complexes with binding energies ranging from 14.5 to 20.1 kJ mol−1.

![Pyridine, 4-[2-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/800/718-24-1.png)
![Pyridine, 4-[2-(4-methylphenyl)ethenyl]-](http://img.cochemist.com/ccimg/800/718-24-1_b.png)




![Pyridine, 4-[2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/800/722-21-4.png)
![Pyridine, 4-[2-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/800/722-21-4_b.png)
![cis-Bicyclo[3.2.0]hept-2-ene](http://img.cochemist.com/ccimg/7100/7095-65-0.png)
![cis-Bicyclo[3.2.0]hept-2-ene](http://img.cochemist.com/ccimg/7100/7095-65-0_b.png)