Co-reporter:Lili Xing, Junwei Lucas Bao, Zhandong Wang, Feng Zhang, and Donald G. Truhlar
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15821-15821
Publication Date(Web):October 12, 2017
DOI:10.1021/jacs.7b08297
Oxygenates with carbonyl and hydroperoxy functional groups are important intermediates that are generated during the autoxidation of organic compounds in the atmosphere and during the autoignition of transport fuels. In the troposphere, the degradation of carbonyl hydroperoxides leads to low-vapor-pressure polyfunctional species that may precipitate in clouds and fog droplets or to the formation of secondary organic aerosols (SOAs). In combustion, the fate of carbonyl hydroperoxides is important for the performance of advanced combustion engines, especially for autoignition. A key fate of the carbonyl hydroperoxides is reaction with OH radicals, for which kinetics data are experimentally unavailable. Here, we study 4-hydroperoxy-2-pentanone (CH3C(═O)CH2CH(OOH)CH3) as a model compound to clarify the kinetics of OH reactions with carbonyl hydroperoxides, in particular H atom abstraction and OH addition reactions. With a combination of electronic structure calculations, we determine previously missing thermochemical data, and with multipath variational transition state theory (MP-VTST), a multidimensional tunneling (MT) approximation, multiple-structure anharmonicity, and torsional potential anharmonicity, we obtained much more accurate rate constants than the ones that can computed by conventional single-structure harmonic transition state theory (TST) and than the empirically estimated rate constants that are currently used in atmospheric and combustion modeling. The roles of various factors in determining the rates are elucidated. The pressure-dependent rate constants for the addition reaction are computed using system-specific quantum RRK theory. The calculated temperature range is 298–2400 K, and the pressure range is 0.01–100 atm. The accurate thermodynamic and kinetics data determined in this work are indispensable in the global modeling of SOAs in atmospheric science and in the detailed understanding and prediction of ignition properties of hydrocarbons and alternative fuels.
Co-reporter:Peng Zhang, Shuang Li, Yingdi Wang, Weiqi Ji, ... Feng Zhang
Combustion and Flame 2017 Volume 183(Volume 183) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.combustflame.2017.05.006
This paper proposes a method to measure the reaction rate constants in kinetically-simple pyrolysis systems using rapid compression machine (RCM) and fast sampling. The method involves first performing sensitivity analysis based on a reasonable kinetic model to identify the species dominated by a target reaction. Then the time-resolved species concentration profiles are measured in RCM experiments using fast sampling and gas chromatography. Finally, the pre-assigned pre-exponential factor and the activation energy are optimized by an iterative fitting procedure, in which the entire temperature profile derived from the pressure history is taken into account. In order to validate this method, the rate constant of the reaction CH3OCHO (methyl formate, MF) => CH3OH + CO (R1) was determined by measuring the CO concentration over 948–1112 K at 30 bar, obtaining the rate expressionkR1/s−1=3.04×1013exp(−30968K/T), which is consistent with previous theoretical and experimental studies. The rate constant of the reaction CH3OCOOCH3 (dimethyl carbonate, DMC) => CH3OCH3 (dimethyl ether, DME) + CO2 (R2) was then studied by measuring the time-resolved DME concentration over 994–1068 K at 30 bar. The measured rate expression of kR2/s−1=2.02×1013exp(−34248K/T)with an uncertainty of ±30% agrees well with the RRKM/Master Equation calculation based on a high-level quantum chemical potential energy surface.
Co-reporter:Can Huang, Bin Yang, Feng Zhang
Combustion and Flame 2017 Volume 184(Volume 184) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.combustflame.2017.06.003
1,3-Butadiene (13C4H6, CH2CHCHCH2) is of great importance as a ubiquitous intermediate in hydrocarbon combustion. Besides its toxic nature, it is involved in the formation of major soot precursors such as C3H3 and n-/i-C4H5. In the present study, the initiation mechanism of 1,3-butadiene combustion has been systematically investigated. It includes the thermal decomposition and mutual isomerization on the C4H6 potential energy surface (PES), the H-assisted isomerization reaction on the C4H7 PES and the H addition-elimination and H abstraction reactions. The temperature- and pressure-dependent rate coefficients for the first two categories of reactions were computed extensively while the H-addition and H-abstraction reactions were adopted from literature. The updated initiation mechanism was then incorporated into Aramco Mech 2.0 to reveal the initiation pathways in 1,3-butadiene pyrolysis and premixed flame. The previously missing “well-skipping” path 13C4H6CH3 + C3H3 is found to be important in 1,3-butadiene pyrolysis. The H-assisted isomerization has a minor contribution to conversion between 1,3- and 1,2-butadiene, while the similar reactions were suggested to play important roles in fulvene/benzene and allene/propyne interconversion. Using the updated model that includes the above pressure-dependent reactions, the effect of the well-skipping paths on the formation of several well-recognized soot precursors (such as C3H3, C4H5) are discussed.
Co-reporter:Lili Xing, Lidong Zhang, Feng Zhang, Jun Jiang
Combustion and Flame 2017 Volume 182(Volume 182) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.combustflame.2017.03.025
To improve the understanding of low temperature oxidation of methyl cyclohexane, the potential energy surfaces for O2 addition to two of its radicals (tcy-C6H10(*)CH3 with a tertiary radical site and ortho-cy-C6H10(*)CH3 with a secondary radical site in the ortho position to the methyl substitution) have been investigated by high level quantum chemical calculations. The reaction kinetics was studied by the ab initio transition state theory based on master equation methodology. The relationship between low temperature oxidation reactivity and molecular structures was explored, typically in terms of the barrier heights of 1,5 H-shift for peroxy radicals. The computed phenomenological rate constants reveal that the ROO stabilization dominates the fates of two methyl cyclohexyl radicals reacting with O2 at pressures higher than 1 atm over the studied temperature range. The balance between ROO stabilization, HO2 elimination, QOOH stabilization, and OH formation was further revealed by branching ratios of these four types of reactions. This study extends kinetic data for cyclic alkanes oxidation in a wide range of pressure and temperature.
Co-reporter:Zhaohui Wang, Lidong Zhang, and Feng Zhang
The Journal of Physical Chemistry A 2014 Volume 118(Issue 34) pp:6741-6748
Publication Date(Web):August 4, 2014
DOI:10.1021/jp503325p
Homoallylic/homobenzylic radicals refer to typical radicals with the radical site located at the β position from the vinyl/phenyl group. These radicals are largely involved in combustion systems, such as the pyrolysis or oxidation of alkenes, cycloalkanes, and aromatics. The 1,2-vinyl/phenyl migration via two steps (cyclization/fission) is a peculiar reaction type for the homoallylic/homobenzylic radicals, entitled homoallylic/homobenzylic rearrangement, which has been studied by theoretical calculations including the Hirshfeld atomic charge analysis in the present work. With the help of rate constant calculations, the competition between this reaction channel and other possible pathways under combustion temperatures (500–2000 K) were evaluated. Analogous 1,3- and 1,4-vinyl/phenyl migration reactions for similar radicals with the radical sites located at the γ and δ positions from the vinyl/phenyl group were also computed. The results indicate that the 1,2-vinyl/phenyl migration is particularly important for the kinetics of unimolecular reactions of homoallylic radicals under 1500 K; nevertheless, it still has noticeable contribution at higher temperature. For those radicals with the radical site at the γ or δ positions, the respective 1,3- or 1,4-vinyl/phenyl migration channel plays an insignificant role under combustion conditions.
Co-reporter:Liangyuan Jia, Jiuzhong Yang, Lidong Zhang, Feng Zhang, Fei Qi, Haiyan Fan, Jibao Cai
Journal of Analytical and Applied Pyrolysis 2013 100() pp: 237-244
Publication Date(Web):
DOI:10.1016/j.jaap.2013.01.002
Co-reporter:Long Zhao, Lili Ye, Feng Zhang, and Lidong Zhang
The Journal of Physical Chemistry A 2012 Volume 116(Issue 37) pp:9238-9244
Publication Date(Web):August 21, 2012
DOI:10.1021/jp305885s
Pentanol is one of the promising “next generation” alcohol fuels with high energy density and low hygroscopicity. In the present work, dominant reaction channels of thermal decomposition of three isomers of pentanol: 1-pentanol, 2-methyl-1-butanol, and 3-methyl-1-butanol were investigated by CBS-QB3 calculations. Subsequently, the temperature- and pressure-dependent rate constants for these channels were computed by RRKM/master equation simulations. The difference between the thermal decomposition behavior of pentanol and butanol were discussed, while butanol as another potential alternative alcohol fuel has been extensively studied both experimentally and theoretically. Rate constants of barrierless bond dissociation reactions of pentanol isomers were treated by the variational transition state theory. The comparison between various channels revealed that the entropies of variational transition states significantly impact the rate constants of pentanol decomposition reactions. This work provides sound quality kinetic data for major decomposition channels of three pentanol isomers in the temperature range of 800–2000 K with pressure varying from 7.6 to 7.6 × 104 Torr, which might be valuable for developing detailed kinetic models for pentanol combustion.
Co-reporter:Can Huang, Bin Yang, Feng Zhang
Combustion and Flame (July 2017) Volume 181() pp:100-109
Publication Date(Web):1 July 2017
DOI:10.1016/j.combustflame.2017.01.031
Butene (C4H8), the smallest alkene possessing both branched and straight-chain isomers, is usually an important intermediate with relatively high concentration in the combustion of hydrocarbons and oxygenated fuels. In the present study, the kinetics of thermal isomerization, decomposition and chemical activation reactions of three typical butenyl isomers, i.e. nC4H7 (CH2=CHCH2ĊH2), saxC4H7 (CH2=CHĊHCH3) and iC4H7 (CH2=C(CH3)ĊH2), were systematically investigated by theoretical calculations. High-level ab initio calculations coupled with the RRKM/master equation method were used to compute the temperature and pressure dependent rate coefficients. The results show that the existence of vinylic CC bond in iC4H7 largely impedes its decomposition rate. The transformation from iC4H7 to straight-chain C4H7 is kinetically unfavorable due to the high strain energy of the 3-membered ring structure of the isomerization transition state. Furthermore, the calculated rate coefficients were incorporated into USC Mech II and Aramco Mech 2.0 to examine the impact of our computed pressure-dependent kinetics on model predictions. Although the simulation results demonstrate limited improvement on ignition delay time and laminar flame speed of butene, substantial changes are observed for the mole fractions of important intermediate species, e.g., allene and 1,3-butediene, in butene pyrolysis. Modified Arrhenius representations of the calculated rate constants are given and should be used in combustion modeling.
Co-reporter:Huiting Bian, Zhandong Wang, Jinhua Sun, Feng Zhang
Proceedings of the Combustion Institute (2017) Volume 36(Issue 1) pp:237-244
Publication Date(Web):1 January 2017
DOI:10.1016/j.proci.2016.07.049
With the “strain-free” cyclic structure, cyclohexane and alkyl cyclohexanes (and their radicals) have various conformers (e.g. chair, boat, and twist etc.) by pseudorotation of the alkyl ring. Noting that different conformers will undergo different types of H-migration reactions, the mechanism of conformational change may impact the distribution of cyclohexyl and the branched cyclohexyl radical isomers during cyclohexane and alkyl cyclohexanes combustion. Consequently, it will influence the formation of subsequent decomposition products. In this work, the conformational inversion-topomerization mechanism and H-migration reactions for six ethylcyclohexyl radical isomers were systematically studied by ab initio calculations and the transition state theory. The updated sub-mechanism of these conformational changes is incorporated into an ethylcyclohexane pyrolysis model. By comparing the simulated results of the “complete” model including the sub-mechanism of conformational changes and the simplified model ignoring these processes, the effect of inversion-topomerization mechanism on the relative concentrations of various ethylcyclohexyl radicals and the formation of subsequent decomposition products were revealed.
Co-reporter:Lili Xing, Feng Zhang, Lidong Zhang
Proceedings of the Combustion Institute (2017) Volume 36(Issue 1) pp:179-186
Publication Date(Web):1 January 2017
DOI:10.1016/j.proci.2016.08.050
The oxidation mechanism of cyclohexylmethyl radical (cy-C6H11CH2), a prototypical alkyl-substituted cycloalkyl radical, has been investigated by high level quantum chemical calculations, and the chemical kinetics was studied by the variational transition state theory and the Rice–Ramsperger–Kassel–Marcus/Master-Equation theory. The relationship between molecular structure and reactivity was explored for the cyclohexylmethyl peroxy radical, which was also compared with chain-like alkyl peroxy radicals. It is shown that the 1,5 H-shift reaction is more competitive than the 1,6 H-shift for the cy-C6H11CH2OO radical, and at high temperatures the concerted elimination reaction channel forming HO2 and methylenecyclohexane bimolecular products becomes more important. Comparing with cyclohexane, the presence of a methyl group in cyclic alkanes prompts the 1,5-H shift of the corresponding peroxy radical and accelerates the overall low temperature chain branching reaction rate. The current study extends kinetic data involved in cy-C6H11CH2 oxidation including cy-C6H11CH2 and O2 recombination, subsequent isomerization and dissociation of the cy-C6H11CH2OO radical over wide pressure and temperature range.
Co-reporter:Feng Zhang, André Nicolle, Lili Xing, Stephen J. Klippenstein
Proceedings of the Combustion Institute (2017) Volume 36(Issue 1) pp:169-177
Publication Date(Web):1 January 2017
DOI:10.1016/j.proci.2016.06.021
The addition of molecular oxygen to hydrocarbon radicals yields peroxy radicals (ROO), which are crucial species in both atmospheric and combustion chemistry. For aromatic radicals there is little known about the recombination kinetics, especially for the high temperatures of relevance to combustion. Here, we have employed direct CASPT2 based variable reaction coordinate transition state theory to predict the high pressure recombination rates for four prototypical aromatic hydrocarbon radicals: phenyl, benzyl, 1-naphthyl, and 2-naphthyl. The variation in the predicted rates is discussed in relation to their molecular structure. The predicted rate coefficients are in reasonably satisfactory agreement with the limited experimental data and are expected to find utility in chemical modeling studies of PAH growth and oxidation.
Co-reporter:Zhandong Wang, Huiting Bian, Yu Wang, Lidong Zhang, ... Fei Qi
Proceedings of the Combustion Institute (2015) Volume 35(Issue 1) pp:367-375
Publication Date(Web):1 January 2015
DOI:10.1016/j.proci.2014.05.119
To get a better understanding of the combustion chemistry of cycloalkanes with long side chain, the pyrolysis of ethylcyclohexane (ECH) was studied in a flow reactor at atmospheric pressure. The pyrolysis species were analyzed by two methods, synchrotron vacuum ultraviolet photoionization mass spectrometry and gas chromatography. Dozens of species were identified and quantified, including lots of isomers. The emphasis of this study is to investigate the primary decomposition of ECH, including its initial decomposition, isomerization, and further reactions of the cyclic C8H15 radicals formed from the H-abstraction of ECH. The observation of C8H16 alkene indicates the existence of ring-opening isomerization reaction of ECH. The ring-opening isomerization reaction of cyclic C8H15 radicals produces alkenyl radicals, whose further decomposition constitutes the various chain and branched intermediates in ECH pyrolysis. The formation of isoprene and vinylcyclopentane is discussed, which highlights isomerization reactions of radical addition on the double bond of alkenyl radicals, such as oct-6-en-1-yl and oct-5-en-1-yl radicals. The theoretical calculation on the reaction pathways of oct-5-en-1-yl radical also shows that its internal H-migration pathway via eight-membered ring might be competitive to the one via five-membered ring. On the other hand, the decomposition of cyclic C8H15 radicals causes the formation of cyclic intermediates, i.e. C8H14 alkenes, methylenecyclohexane and cyclohexene, which are potential aromatic precursors.








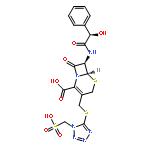
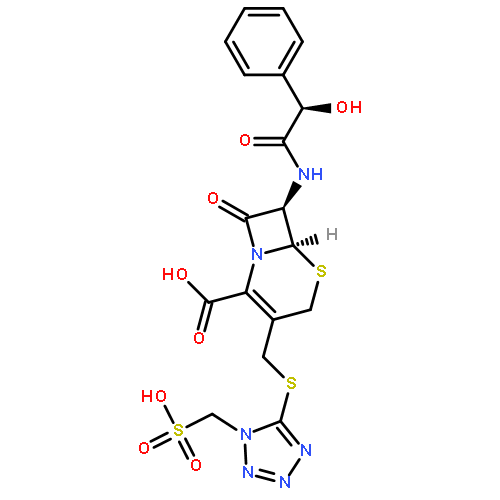
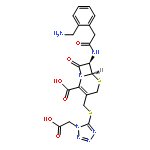





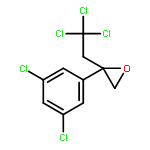
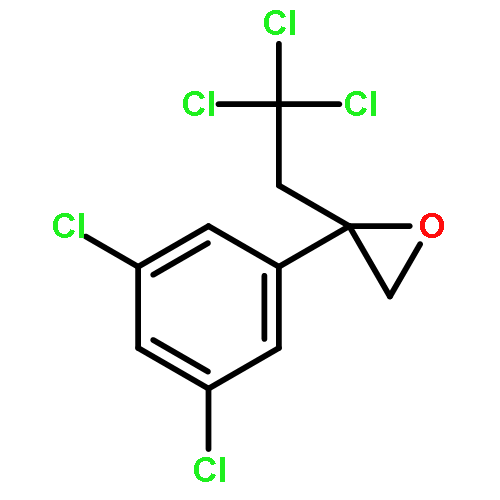
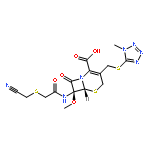
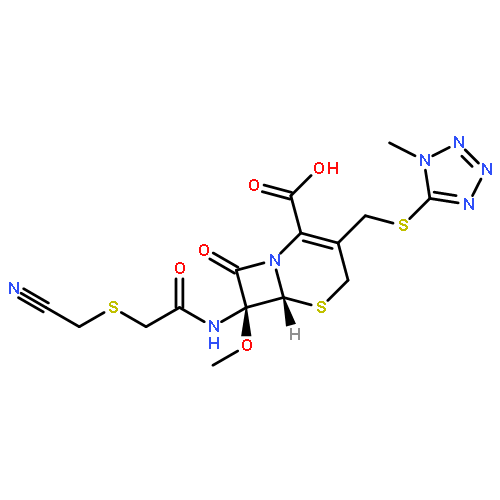
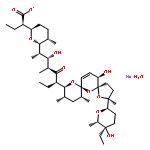


![2-[3-(TRIFLUOROMETHYL)PHENYL]MORPHOLINE](http://img.cochemist.com/ccimg/54800/54774-45-7.png)
![2-[3-(TRIFLUOROMETHYL)PHENYL]MORPHOLINE](http://img.cochemist.com/ccimg/54800/54774-45-7_b.png)





![Furan,[(methylthio)methyl]-](http://img.cochemist.com/ccimg/40300/40228-18-0.png)
![Furan,[(methylthio)methyl]-](http://img.cochemist.com/ccimg/40300/40228-18-0_b.png)
![N-[4-[(5-METHYL-1,3,4-THIADIAZOL-2-YL)SULFAMOYL]PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/39800/39719-87-4.png)
![N-[4-[(5-METHYL-1,3,4-THIADIAZOL-2-YL)SULFAMOYL]PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/39800/39719-87-4_b.png)


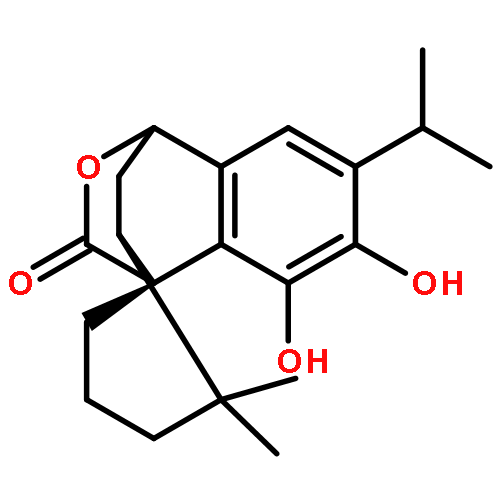
![5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylicacid, 3-(hydroxymethyl)-8-oxo-7-[[2-(4-pyridinylthio)acetyl]amino]-, (6R,7R)-](http://img.cochemist.com/ccimg/38200/38115-21-8.png)
![5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylicacid, 3-(hydroxymethyl)-8-oxo-7-[[2-(4-pyridinylthio)acetyl]amino]-, (6R,7R)-](http://img.cochemist.com/ccimg/38200/38115-21-8_b.png)






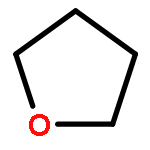
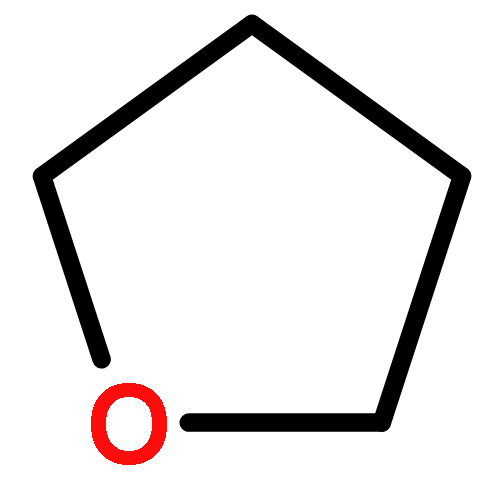

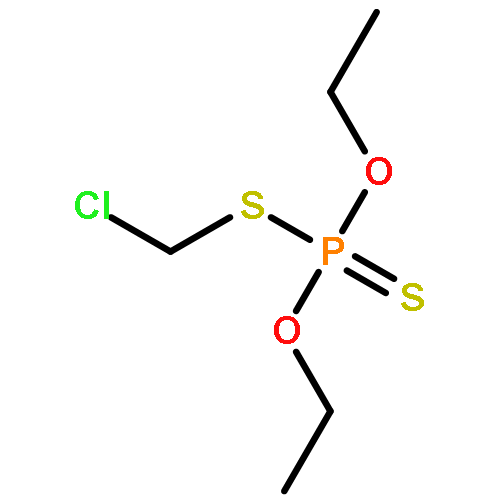


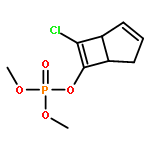
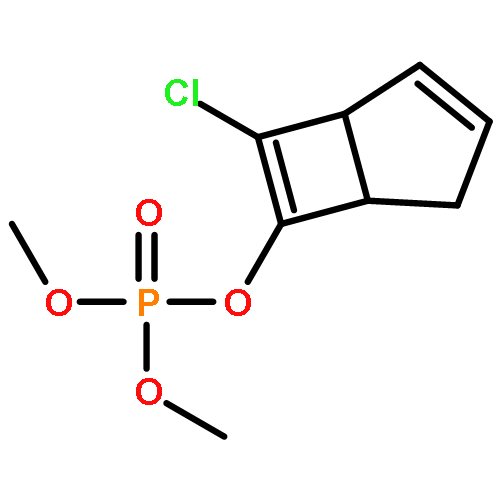
![Phosphorothioic acid,O-[2,5-dichloro-4-(methylthio)phenyl] O,O-diethyl ester](http://img.cochemist.com/ccimg/22000/21923-23-9.png)
![Phosphorothioic acid,O-[2,5-dichloro-4-(methylthio)phenyl] O,O-diethyl ester](http://img.cochemist.com/ccimg/22000/21923-23-9_b.png)


![5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylicacid, 3-[(acetyloxy)methyl]-8-oxo-7-[[(4-pyridinylthio)acetyl]amino]-, (6R,7R)-](http://img.cochemist.com/ccimg/21600/21593-23-7.png)
![5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylicacid, 3-[(acetyloxy)methyl]-8-oxo-7-[[(4-pyridinylthio)acetyl]amino]-, (6R,7R)-](http://img.cochemist.com/ccimg/21600/21593-23-7_b.png)
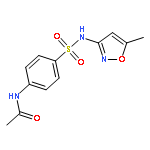
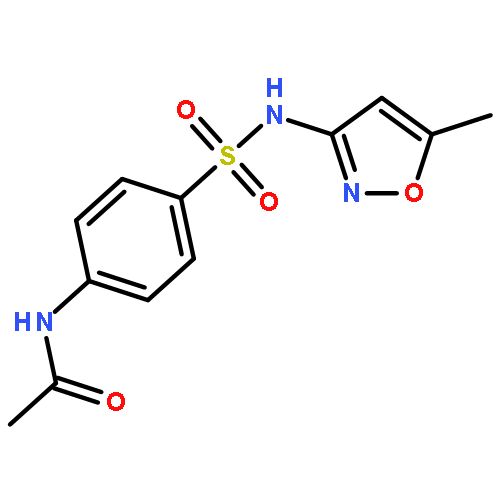
![Acetamide,N-[4-[(2-pyridinylamino)sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/19100/19077-98-6.png)
![Acetamide,N-[4-[(2-pyridinylamino)sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/19100/19077-98-6_b.png)
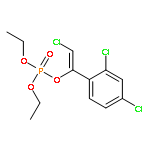

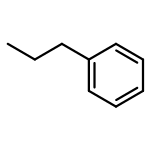
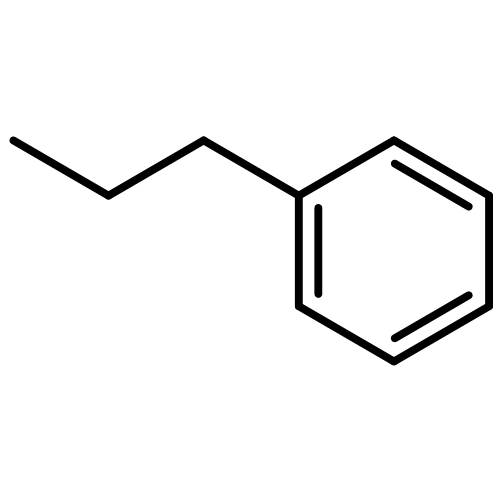
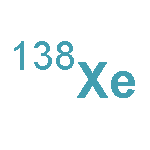









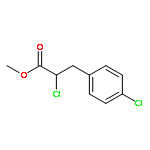






























![Acetamide,N-[[4-(acetylamino)phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/5700/5626-90-4.png)
![Acetamide,N-[[4-(acetylamino)phenyl]sulfonyl]-](http://img.cochemist.com/ccimg/5700/5626-90-4_b.png)
![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6.png)
![(e)-1-(2,4-dihydroxyphenyl)-3-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]prop-2-en-1-one](http://img.cochemist.com/ccimg/5100/5041-81-6_b.png)











![Thiodiphosphoric acid([(HO)2P(S)]2O), OP,OP,OP',OP'-tetrapropyl ester](http://img.cochemist.com/ccimg/3300/3244-90-4.png)
![Thiodiphosphoric acid([(HO)2P(S)]2O), OP,OP,OP',OP'-tetrapropyl ester](http://img.cochemist.com/ccimg/3300/3244-90-4_b.png)


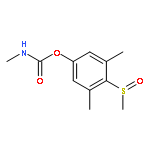
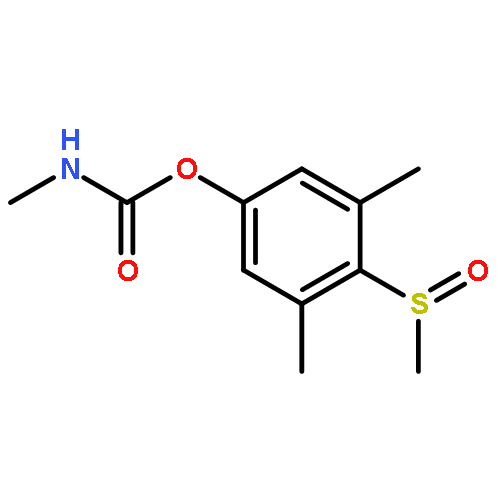


![Phosphorothioic acid,O,O-diethyl S-[(ethylsulfonyl)methyl] ester](http://img.cochemist.com/ccimg/2600/2588-06-9.png)
![Phosphorothioic acid,O,O-diethyl S-[(ethylsulfonyl)methyl] ester](http://img.cochemist.com/ccimg/2600/2588-06-9_b.png)
![Phosphorodithioic acid,O,O-diethyl S-[(ethylsulfonyl)methyl] ester](http://img.cochemist.com/ccimg/2600/2588-04-7.png)
![Phosphorodithioic acid,O,O-diethyl S-[(ethylsulfonyl)methyl] ester](http://img.cochemist.com/ccimg/2600/2588-04-7_b.png)
![Phosphorodithioic acid,O,O-diethyl S-[2-(ethylsulfinyl)ethyl] ester](http://img.cochemist.com/ccimg/2500/2497-07-6.png)
![Phosphorodithioic acid,O,O-diethyl S-[2-(ethylsulfinyl)ethyl] ester](http://img.cochemist.com/ccimg/2500/2497-07-6_b.png)
![Phosphorothioic acid,O,O-diethyl S-[2-(ethylsulfonyl)ethyl] ester](http://img.cochemist.com/ccimg/2500/2496-91-5.png)
![Phosphorothioic acid,O,O-diethyl S-[2-(ethylsulfonyl)ethyl] ester](http://img.cochemist.com/ccimg/2500/2496-91-5_b.png)




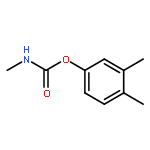
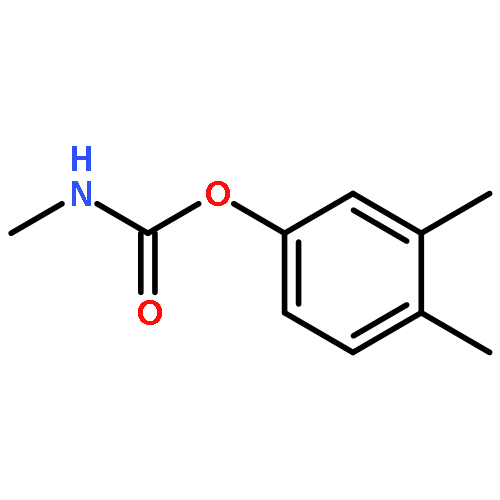
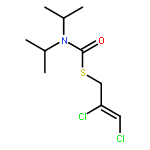

![Phosphorodithioic acid,O,O-diethyl S-[2-[(1-methylethyl)amino]-2-oxoethyl] ester](http://img.cochemist.com/ccimg/2300/2275-18-5.png)
![Phosphorodithioic acid,O,O-diethyl S-[2-[(1-methylethyl)amino]-2-oxoethyl] ester](http://img.cochemist.com/ccimg/2300/2275-18-5_b.png)
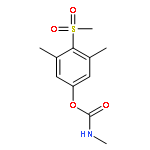
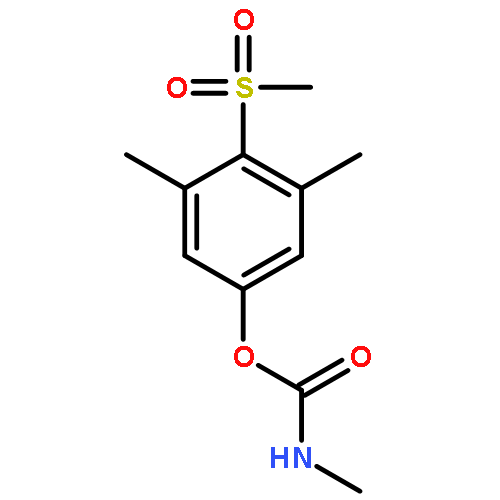




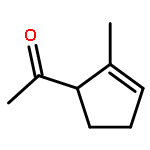


![5-(Bromomethyl)-1,2,3,4,7,7-hexachlorobicyclo[2.2.1]hept-2-ene](http://img.cochemist.com/ccimg/1800/1715-40-8.png)
![5-(Bromomethyl)-1,2,3,4,7,7-hexachlorobicyclo[2.2.1]hept-2-ene](http://img.cochemist.com/ccimg/1800/1715-40-8_b.png)




![1,4-Naphthalenedione,2-methyl-3-[(2E,6E,10E,14E)-3,7,11,15,19-pentamethyl-2,6,10,14,18-eicosapentaen-1-yl]-](http://img.cochemist.com/ccimg/1200/1182-68-9.png)
![1,4-Naphthalenedione,2-methyl-3-[(2E,6E,10E,14E)-3,7,11,15,19-pentamethyl-2,6,10,14,18-eicosapentaen-1-yl]-](http://img.cochemist.com/ccimg/1200/1182-68-9_b.png)
![Phosphoric acid,4-[(dimethylamino)sulfonyl]phenyl dimethyl ester](http://img.cochemist.com/ccimg/1000/960-25-8.png)
![Phosphoric acid,4-[(dimethylamino)sulfonyl]phenyl dimethyl ester](http://img.cochemist.com/ccimg/1000/960-25-8_b.png)












![(2s)-7-hydroxy-2-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/600/551-15-5.png)
![(2s)-7-hydroxy-2-[4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyphenyl]-2,3-dihydrochromen-4-one](http://img.cochemist.com/ccimg/600/551-15-5_b.png)













![Phosphorothioic acid,O,O-diethyl O-[2-(ethylthio)ethyl] ester](http://img.cochemist.com/ccimg/300/298-03-3.png)
![Phosphorothioic acid,O,O-diethyl O-[2-(ethylthio)ethyl] ester](http://img.cochemist.com/ccimg/300/298-03-3_b.png)





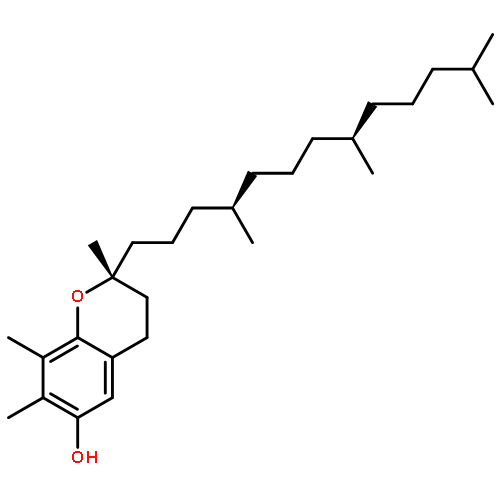
![2H-1-Benzopyran-6-ol,3,4-dihydro-2,5,8-trimethyl-2-[(4R,8R)-4,8,12-trimethyltridecyl]-, (2R)-rel-](http://img.cochemist.com/ccimg/200/148-03-8.png)
![2H-1-Benzopyran-6-ol,3,4-dihydro-2,5,8-trimethyl-2-[(4R,8R)-4,8,12-trimethyltridecyl]-, (2R)-rel-](http://img.cochemist.com/ccimg/200/148-03-8_b.png)
![Acetamide,N-[4-[(2-pyrimidinylamino)sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/200/127-74-2.png)
![Acetamide,N-[4-[(2-pyrimidinylamino)sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/200/127-74-2_b.png)
![Acetamide,N-[4-[[(4-methyl-2-pyrimidinyl)amino]sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/200/127-73-1.png)
![Acetamide,N-[4-[[(4-methyl-2-pyrimidinyl)amino]sulfonyl]phenyl]-](http://img.cochemist.com/ccimg/200/127-73-1_b.png)












![5-((2-(2-Butoxyethoxy)ethoxy)methyl)-6-propylbenzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/100/51-03-6.png)
![5-((2-(2-Butoxyethoxy)ethoxy)methyl)-6-propylbenzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/100/51-03-6_b.png)