Co-reporter:Michael L. Baker, Michael W. Mara, James J. Yan, Keith O. Hodgson, Britt Hedman, Edward I. Solomon
Coordination Chemistry Reviews 2017 Volume 345(Volume 345) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.ccr.2017.02.004
•Directly probe metal oxidation state, geometry and coordination number.•Quantify differential orbital covalency.•Spectroscopic access to highly-covalent sites, including hemes.•Application of resonant inelastic X-ray scattering to bioinorganic sites.Continual advancements in the development of synchrotron radiation sources have resulted in X-ray based spectroscopic techniques capable of probing the electronic and structural properties of numerous systems. This review gives an overview of the application of metal K-edge and L-edge X-ray absorption spectroscopy (XAS), as well as Kα resonant inelastic X-ray scattering (RIXS), to the study of electronic structure in transition metal sites with emphasis on experimentally quantifying 3d orbital covalency. The specific sensitivities of K-edge XAS, L-edge XAS, and RIXS are discussed emphasizing the complementary nature of the methods. L-edge XAS and RIXS are sensitive to mixing between 3d orbitals and ligand valence orbitals, and to the differential orbital covalency (DOC), that is, the difference in the covalencies for different symmetry sets of the d orbitals. Both L-edge XAS and RIXS are highly sensitive to and enable separation of σ and π donor bonding and π back bonding contributions to bonding. Applying ligand field multiplet simulations, including charge transfer via valence bond configuration interactions, DOC can be obtained for direct comparison with density functional theory calculations and to understand chemical trends. The application of RIXS as a probe of frontier molecular orbitals in a heme enzyme demonstrates the potential of this method for the study of metal sites in highly covalent coordination sites in bioinorganic chemistry.
Co-reporter:James J. YanMargarita A. Gonzales, Pradip K. Mascharak, Britt HedmanKeith O. Hodgson, Edward I. Solomon
Journal of the American Chemical Society 2016 Volume 139(Issue 3) pp:1215-1225
Publication Date(Web):December 22, 2016
DOI:10.1021/jacs.6b11260
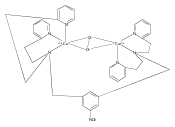
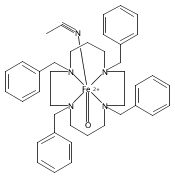
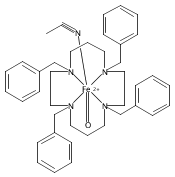
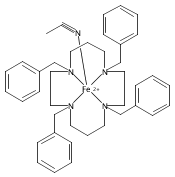
NO is a classic non-innocent ligand, and iron nitrosyls can have different electronic structure descriptions depending on their spin state and coordination environment. These highly covalent ligands are found in metalloproteins and are also used as models for Fe–O2 systems. This study utilizes iron L-edge X-ray absorption spectroscopy (XAS), interpreted using a valence bond configuration interaction multiplet model, to directly experimentally probe the electronic structure of the S = 0 {FeNO}6 compound [Fe(PaPy3)NO]2+ (PaPy3 = N,N-bis(2-pyridylmethyl)amine-N-ethyl-2-pyridine-2-carboxamide) and the S = 0 [Fe(PaPy3)CO]+ reference compound. This method allows separation of the σ-donation and π-acceptor interactions of the ligand through ligand-to-metal and metal-to-ligand charge-transfer mixing pathways. The analysis shows that the {FeNO}6 electronic structure is best described as FeIII–NO(neutral), with no localized electron in an NO π* orbital or electron hole in an Fe dπ orbital. This delocalization comes from the large energy gap between the Fe–NO π-bonding and antibonding molecular orbitals relative to the exchange interactions between electrons in these orbitals. This study demonstrates the utility of L-edge XAS in experimentally defining highly delocalized electronic structures.
Co-reporter:Aaron W. Fay;Markus W. Ribbe;Chi Chung Lee;Michael A. Blank;Johannes G. Rebelein;Britt Hedman;Yilin Hu
PNAS 2016 Volume 113 (Issue 34 ) pp:9504-9508
Publication Date(Web):2016-08-23
DOI:10.1073/pnas.1609574113
NifEN is a biosynthetic scaffold for the cofactor of Mo-nitrogenase (designated the M-cluster). Previous studies have revealed
the sequence and structural homology between NifEN and NifDK, the catalytic component of nitrogenase. However, direct proof
for the functional homology between the two proteins has remained elusive. Here we show that, upon maturation of a cofactor
precursor (designated the L-cluster) on NifEN, the cluster species extracted from NifEN is spectroscopically equivalent and
functionally interchangeable with the native M-cluster extracted from NifDK. Both extracted clusters display nearly indistinguishable
EPR features, X-ray absorption spectroscopy/extended X-ray absorption fine structure (XAS/EXAFS) spectra and reconstitution
activities, firmly establishing the M-cluster–bound NifEN (designated NifENM) as the only protein other than NifDK to house the unique nitrogenase cofactor. Iron chelation experiments demonstrate a
relocation of the cluster from the surface to its binding site within NifENM upon maturation, which parallels the insertion of M-cluster into an analogous binding site in NifDK, whereas metal analyses
suggest an asymmetric conformation of NifENM with an M-cluster in one αβ-half and an empty cluster-binding site in the other αβ-half, which led to the proposal of a stepwise
assembly mechanism of the M-cluster in the two αβ-dimers of NifEN. Perhaps most importantly, NifENM displays comparable ATP-independent substrate-reducing profiles to those of NifDK, which establishes the M-cluster–bound
αβ-dimer of NifENM as a structural and functional mimic of one catalytic αβ-half of NifDK while suggesting the potential of this protein as
a useful tool for further investigations of the mechanistic details of nitrogenase.
Co-reporter:Thomas Kroll ; Ryan G. Hadt ; Samuel A. Wilson ; Marcus Lundberg ; James J. Yan ; Tsu-Chien Weng ; Dimosthenis Sokaras ; Roberto Alonso-Mori ; Diego Casa ; Mary H. Upton ; Britt Hedman ; Keith O. Hodgson ;Edward I. Solomon
Journal of the American Chemical Society 2014 Volume 136(Issue 52) pp:18087-18099
Publication Date(Web):December 4, 2014
DOI:10.1021/ja5100367
Axial Cu–S(Met) bonds in electron transfer (ET) active sites are generally found to lower their reduction potentials. An axial S(Met) bond is also present in cytochrome c (cyt c) and is generally thought to increase the reduction potential. The highly covalent nature of the porphyrin environment in heme proteins precludes using many spectroscopic approaches to directly study the Fe site to experimentally quantify this bond. Alternatively, L-edge X-ray absorption spectroscopy (XAS) enables one to directly focus on the 3d-orbitals in a highly covalent environment and has previously been successfully applied to porphyrin model complexes. However, this technique cannot be extended to metalloproteins in solution. Here, we use metal K-edge XAS to obtain L-edge like data through 1s2p resonance inelastic X-ray scattering (RIXS). It has been applied here to a bis-imidazole porphyrin model complex and cyt c. The RIXS data on the model complex are directly correlated to L-edge XAS data to develop the complementary nature of these two spectroscopic methods. Comparison between the bis-imidazole model complex and cyt c in ferrous and ferric oxidation states show quantitative differences that reflect differences in axial ligand covalency. The data reveal an increased covalency for the S(Met) relative to N(His) axial ligand and a higher degree of covalency for the ferric states relative to the ferrous states. These results are reproduced by DFT calculations, which are used to evaluate the thermodynamics of the Fe–S(Met) bond and its dependence on redox state. These results provide insight into a number of previous chemical and physical results on cyt c.
Co-reporter:Munzarin F. Qayyum ; Ritimukta Sarangi ; Kiyoshi Fujisawa ; T. Daniel P. Stack ; Kenneth D. Karlin ; Keith O. Hodgson ; Britt Hedman ;Edward I. Solomon
Journal of the American Chemical Society 2013 Volume 135(Issue 46) pp:17417-17431
Publication Date(Web):October 8, 2013
DOI:10.1021/ja4078717
The hydroxylation of aromatic substrates catalyzed by coupled binuclear copper enzymes has been observed with side-on-peroxo-dicopper(II) (P) and bis-μ-oxo-dicopper(III) (O) model complexes. The substrate-bound-O intermediate in [Cu(II)2(DBED)2(O)2]2+ (DBED = N,N′-di-tert-butyl-ethylenediamine) was shown to perform aromatic hydroxylation. For the [Cu(II)2(NO2-XYL)(O2)]2+ complex, only a P species was spectroscopically observed. However, it was not clear whether this O–O bond cleaves to proceed through an O-type structure along the reaction coordinate for hydroxylation of the aromatic xylyl linker. Accurate evaluation of these reaction coordinates requires reasonable quantitative descriptions of the electronic structures of the P and O species. We have performed Cu L-edge XAS on two well-characterized P and O species to experimentally quantify the Cu 3d character in their ground state wave functions. The lower per-hole Cu character (40 ± 6%) corresponding to higher covalency in the O species compared to the P species (52 ± 4%) reflects a stronger bonding interaction of the bis-μ-oxo core with the Cu(III) centers. DFT calculations show that 10–20% Hartree–Fock (HF) mixing for P and ∼38% for O species are required to reproduce the Cu–O bonding; for the P species this HF mixing is also required for an antiferromagnetically coupled description of the two Cu(II) centers. B3LYP (with 20% HF) was, therefore, used to calculate the hydroxylation reaction coordinate of P in [Cu(II)2(NO2-XYL)(O2)]2+. These experimentally calibrated calculations indicate that the electrophilic attack on the aromatic ring does not involve formation of a Cu(III)2(O2–)2 species. Rather, there is direct electron donation from the aromatic ring into the peroxo σ* orbital of the Cu(II)2(O22–) species, leading to concerted C–O bond formation with O–O bond cleavage. Thus, species P is capable of direct hydroxylation of aromatic substrates without the intermediacy of an O-type species.
Co-reporter:Samuel A. Wilson ; Thomas Kroll ; Richard A. Decreau ; Rosalie K. Hocking ; Marcus Lundberg ; Britt Hedman ; Keith O. Hodgson ;Edward I. Solomon
Journal of the American Chemical Society 2012 Volume 135(Issue 3) pp:1124-1136
Publication Date(Web):December 22, 2012
DOI:10.1021/ja3103583
The electronic structure of the Fe–O2 center in oxy-hemoglobin and oxy-myoglobin is a long-standing issue in the field of bioinorganic chemistry. Spectroscopic studies have been complicated by the highly delocalized nature of the porphyrin, and calculations require interpretation of multideterminant wave functions for a highly covalent metal site. Here, iron L-edge X-ray absorption spectroscopy, interpreted using a valence bond configuration interaction multiplet model, is applied to directly probe the electronic structure of the iron in the biomimetic Fe–O2 heme complex [Fe(pfp)(1-MeIm)O2] (pfp (“picket fence porphyrin”) = meso-tetra(α,α,α,α-o-pivalamidophenyl)porphyrin or TpivPP). This method allows separate estimates of σ-donor, π-donor, and π-acceptor interactions through ligand-to-metal charge transfer and metal-to-ligand charge transfer mixing pathways. The L-edge spectrum of [Fe(pfp)(1-MeIm)O2] is further compared to those of [FeII(pfp)(1-MeIm)2], [FeII(pfp)], and [FeIII(tpp)(ImH)2]Cl (tpp = meso-tetraphenylporphyrin) which have FeIIS = 0, FeIIS = 1, and FeIIIS = 1/2 ground states, respectively. These serve as references for the three possible contributions to the ground state of oxy-pfp. The Fe–O2 pfp site is experimentally determined to have both significant σ-donation and a strong π-interaction of the O2 with the iron, with the latter having implications with respect to the spin polarization of the ground state.
Co-reporter:Rosalie K. Hocking ; Serena DeBeer George ; Zeev Gross ; F. Ann Walker ; Keith O. Hodgson ; Britt Hedman ;Edward I. Solomon
Inorganic Chemistry 2009 Volume 48(Issue 4) pp:1678-1688
Publication Date(Web):January 16, 2009
DOI:10.1021/ic802248t
Corrole is a tetrapyrrolic macrocycle that has one carbon atom less than a porphyrin. The ring contraction reduces the symmetry from D4h to C2v, changes the electronic structure of the heterocycle, and leads to a smaller central cavity with three protons rather than the two of a porphyrin. The differences between ferric corroles and porphyrins lead to a number of differences in reactivity including increased axial ligand lability and a tendency to form 5-coordinate complexes. The electronic structure origin of these differences has been difficult to study experimentally as the dominant porphyrin/corrole π → π* transitions obscure the electronic transitions of the metal. Recently, we have developed a methodology that allows for the interpretation of the multiplet structure of Fe L-edges in terms of differential orbital covalency (i.e., the differences in mixing of the metal d orbitals with the ligand valence orbitals) using a valence bond configuration interaction model. Herein, we apply this methodology, combined with a ligand field analysis of the Fe K pre-edge to a low-spin ferric corrole, and compare it to a low-spin ferric porphyrin. The experimental results combined with DFT calculations show that the contracted corrole is both a stronger σ donor and a very anisotropic π donor. These differences decrease the bonding interactions with axial ligands and contribute to the increased axial ligand lability and reactivity of ferric corroles relative to ferric porphyrins.


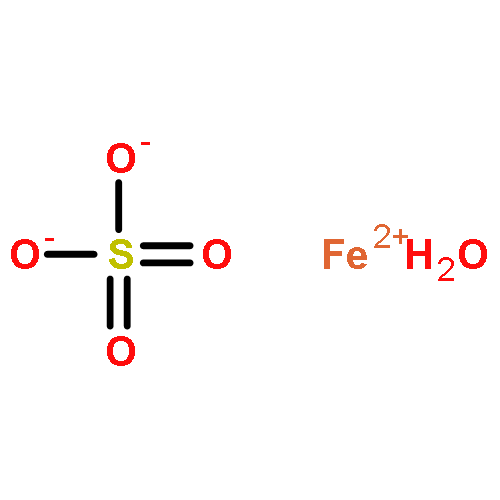
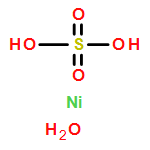
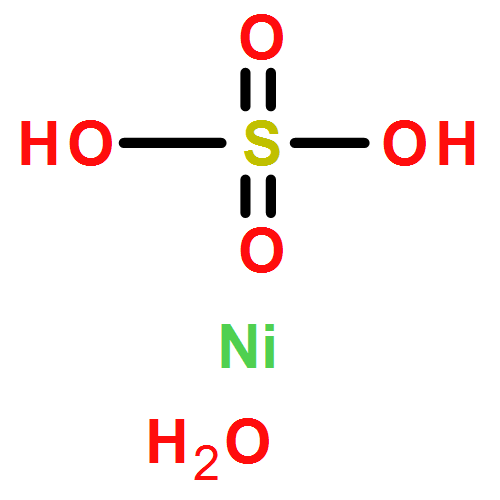
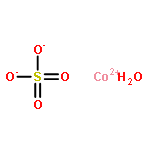





























































































































![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)