Co-reporter:Andong Zhou, Lei Yan, Fangfang Lai, Xiaoguang Chen, Masuo Goto, Kuo-Hsiung Lee, Zhiyan Xiao
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 15(Issue 15) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.bmcl.2017.06.019
The indolin-2-one core is a privileged structure for antitumor agents, especially kinase inhibitors. Twenty-three novel indolin-2-ones were designed by molecular dissection of the anticancer drug indirubin. Seventeen of them exhibited significant inhibition against the tested cell lines, and two of them (1c and 1h) showed IC50 values at the submicromolar level against HCT-116 cells. Compounds 1c and 2c were also potent inhibitors of the triple-negative breast cancer (TNBC) cell line MDA-MB-231. Flow cytometry was utilized to explore the antitumor mechanism of 1c and 2c with MDA-MB-231 cells, and distinct effects were observed on 2c. Furthermore, immunocytochemical examination of 1c suggested a destabilization of microtubules, which was significantly different from the effect of IM, an indirubin derivative.Download high-res image (85KB)Download full-size image
Co-reporter:Kai Xu, Ke Wang, Ying Yang, Ding-An Yan, Li Huang, Chin-Ho Chen, Zhiyan Xiao
European Journal of Medicinal Chemistry 2015 Volume 98() pp:61-68
Publication Date(Web):15 June 2015
DOI:10.1016/j.ejmech.2015.05.023
•Novel linear peptides were designed as non-covalent 20S proteasome inhibitors.•Fifteen linear peptides were synthesized.•Three potent inhibitors selective to the β5-subunit were identified.•Docking study on the most potent compound 6e was performed.A series of linear peptides (6a-6o) were designed based on the known non-covalent 20S proteasome inhibitors TMC-95A and compound 5 via a fragment-based approach. These compounds were synthesized and evaluated against the chymotrypsin-like activity of the human 20S proteasome. Three of them (6d, 6e and 6k) were potent inhibitors with IC50 values at the submicromolar level. These three compounds were selective to the β5-subunit and showed no obvious inhibition against trypsin-like and caspase-like activities of the human 20S proteasome. Docking study of the most potent compound 6e revealed its key interactions with the β5-subunit of the 20S proteasome. These findings have provided a new chemical template for non-covalent proteasome inhibitors, which is ready for further structural optimization to improve both potency and subunit selectivity.A series of linear peptides (6) were designed based on the known non-covalent 20S proteasome inhibitors TMC-95A and compound 5 via a fragment-based approach.
Co-reporter:Jiadi Gao, Cheng Fang, Zhiyan Xiao, Li Huang, Chin-Ho Chen, Li-Ting Wang and Kuo-Hsiung Lee
MedChemComm 2015 vol. 6(Issue 3) pp:444-454
Publication Date(Web):28 Nov 2014
DOI:10.1039/C4MD00412D
Based on a 3D-QSAR pharmacophore derived from a diverse set of known cyclin-dependent kinase 9 (CDK9) inhibitors and a composite pharmacophore extracted from the complex structure of flavopiridol (FVP)-CDK9, thirty novel 5-fluoro-N2,N4-diphenylpyrimidine-2,4-diamine derivatives were designed and synthesized. Initial tests against four tumor cell lines with the sulforhodamine B (SRB) assay identified a series of potent compounds with GI50 values at a lower micromolar or submicromolar level. Most of the highly cytotoxic compounds exhibited potent inhibitory activities against both CDK2/cyclin E1 and CDK9/cyclin T1. Notably, inhibitions against the two enzymes were generally correlated well with the cytotoxicity of these compounds. Appreciable inhibition was also observed for selected compounds in the anti-HIV-1 assay. Docking studies on compounds 6d and 9g provided conducive clues to further structural optimization.
Co-reporter:Lei Yan, Fangfang Lai, Xiaoguang Chen, Zhiyan Xiao
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 11) pp:2447-2451
Publication Date(Web):1 June 2015
DOI:10.1016/j.bmcl.2015.03.066
Indirubin-3′-monoxime (IM) is a potent cyclin-dependent kinase (CDK) inhibitor. Twenty novel IM derivatives were prepared to investigate the structure–activity relationships (SAR) of this compound class. Six compounds showed significant inhibition against both CDK2/cyclin E1 and CDK9/cyclin T1. The most potent compound 7t exhibited IC50 values at submicromolar level. Preliminary SAR trends were suggested and cytotoxicity of these compounds was investigated. Molecular docking studies on compounds 7l and 7t provided conducive clues for further structural optimization.
Co-reporter:Dr. Yan-Bo Tang;Jun-Zheng Liu;Dr. Shu-En Zhang;Xin Du;Feilin Nie;Jin-Ying Tian; Fei Ye;Dr. Kai Huang;Dr. Jin-Ping Hu; Yan Li; Zhiyan Xiao
ChemMedChem 2014 Volume 9( Issue 5) pp:918-921
Publication Date(Web):
DOI:10.1002/cmdc.201400007
Abstract
Protein tyrosine phosphatase 1B (PTP1B) is a promising therapeutic target for type 2 diabetes. Herein, we report the evolution of a previously identified 3-phenylpropanoic acid-based PTP1B inhibitor to an orally active lead compound. A series of 3-phenylpropanoic acid-based PTP1B inhibitors were synthesized, and three of them, 3-(4-(9H-carbazol-9-yl)phenyl)-5-(3,5-di-tert-butyl-4-methoxyphenyl)-5-oxopentanoic acid (9), 3-(4-(9H-carbazol-9-yl)phenyl)-5-(4′-bromo-[1,1′-biphenyl]-4-yl)-5-oxopentanoic acid (10) and 3-(4-(9H-carbazol-9-yl)-2-fluorophenyl)-5-(4-cyclohexylphenyl)-5-oxopentanoic acid (16), showed IC50 values at sub-micromolar level. Further in vivo evaluation indicated the sodium salt of 9 not only exhibited significant insulin-sensitizing and hypoglycemia effects, but also decreased the serum levels of triglyceride and total cholesterol in high-fat-diet-induced insulin resistance model mice. Preliminary in vivo pharmacokinetic studies on the sodium salt of 9 revealed its pharmacokinetic profile after oral administration in rats. These results provide proof-of-concept for the dual effects of PTP1B inhibitors on both glucose and lipid metabolisms.
Co-reporter:Dr. Yan-Bo Tang;Jun-Zheng Liu;Dr. Shu-En Zhang;Xin Du;Feilin Nie;Jin-Ying Tian; Fei Ye;Dr. Kai Huang;Dr. Jin-Ping Hu; Yan Li; Zhiyan Xiao
ChemMedChem 2014 Volume 9( Issue 5) pp:
Publication Date(Web):
DOI:10.1002/cmdc.201490019
Co-reporter:Jun-Zheng Liu, Shu-En Zhang, Feilin Nie, Ying Yang, Yan-Bo Tang, Wenwen Yin, Jin-Ying Tian, Fei Ye, Zhiyan Xiao
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 23) pp:6217-6222
Publication Date(Web):1 December 2013
DOI:10.1016/j.bmcl.2013.10.002
An integrated molecular design strategy combining pharmacophore recognition and scaffold hopping was exploited to discover novel PTP1B inhibitors based on the known PTP1B inhibitor Ertiprotafib. A composite pharmacophore model was proposed from the interaction mode of Ertiprotafib, and 21 diverse molecules from five distinct structural classes were designed and synthesized accordingly. New compounds with considerable inhibition against PTP1B were identified from each series, and the most active compound 3a showed IC50 value of 1.3 μmol L−1 against human recombinant PTP1B. Docking study indicated that the new inhibitors assumed binding modes similar to that of Ertiprotafib.
Co-reporter:Shanfu Wei, Yan-Bo Tang, Huiming Hua, Emika Ohkoshi, Masuo Goto, Li-Ting Wang, Kuo-Hsiung Lee, Zhiyan Xiao
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 14) pp:4056-4060
Publication Date(Web):15 July 2013
DOI:10.1016/j.bmcl.2013.05.061
The natural diterpenoid andrographolide (1) exhibits various biological activities. Seventeen derivatives of 1 were prepared via esterification and etherification of 14-dehydroxy-11,12-didehydroandrographolide (2). Most derivatives demonstrated significant inhibition against tumor cell growth. The most active compounds, 3b and 3c, had GI50 values of 1.46–9.19 μM against A549, DU145, KB and KB-Vin tumor cells. In an immunocytochemical study, treatment with compound 3c disrupted microtubule dynamics in PC-3 cells, but caused no accumulation of metaphase cells, which is a phenotype dissimilar from that of 1. This difference suggests that structural modification of 1 resulted in a shift in the underlying molecular mechanism.
Co-reporter:Yan-Bo Tang, Dianyun Lu, Zheng Chen, Chun Hu, Ying Yang, Jin-Ying Tian, Fei Ye, Li Wu, Zhong-Yin Zhang, Zhiyan Xiao
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 8) pp:2313-2318
Publication Date(Web):15 April 2013
DOI:10.1016/j.bmcl.2013.02.073
Fifteen novel sulfathiazole-related compounds were designed as PTP1B inhibitors based on a previously reported allosteric inhibitor (1) of PTP1B. These compounds were synthesized and evaluated against human recombinant PTP1B. Six compounds (3, 4, 8 and 14–16) exhibited significant inhibitory activity against PTP1B. The most active compound (16) showed IC50 value of 3.2 μM and kinetic analysis indicated that it is a non-competitive inhibitor of PTP1B. Furthermore, compound 16 demonstrated excellent selectivity to PTP1B over other PTPs. It also displayed in vivo insulin sensitizing effect in the insulin resistant mice.
Co-reporter:Jie Li, Hui Ming Hua, Yan Bo Tang, Shipeng Zhang, Emika Ohkoshi, Kuo-Hsiung Lee, Zhi yan Xiao
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 13) pp:4293-4295
Publication Date(Web):1 July 2012
DOI:10.1016/j.bmcl.2012.05.033
Because prior studies have shown inconsistency between structure–activity relationships for podophyllotoxin derivatives as topoisomerase II inhibitors and cytotoxic agents, eight novel podophyllotoxin analogs were synthesized to further explore the effects of structural variations on both A and D rings on activity. The new compounds contain a 4,5-dimethoxy substituted A ring and opened D-ring variants and were prepared by appropriate functional and stereochemical operations at the methylenedioxy group, C7, C8, and C8′. Four compounds (15, 18, 21 and 22) demonstrated noticeable inhibitory activity against A549, DU145, KB and KBvin tumor cells, and the most active compound 18 showed IC50 values less than 10 μg/mL.
Co-reporter:Kai Xu, Zhiyan Xiao, Yan Bo Tang, Li Huang, Chin-Ho Chen, Emika Ohkoshi, Kuo-Hsiung Lee
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 8) pp:2772-2774
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmcl.2012.02.086
Fourteen naphthoquinone derivatives (1–14) were designed based on a putative proteasome inhibitor PI-083. These compounds were synthesized and evaluated against A549, DU145, KB, and KBvin tumor cell lines. Six compounds (2, 4, 8, 9, 10, and 13) showed antiproliferative activities comparable to that of PI-083. Among them, compound 8 was confirmed as a 20S proteasome inhibitor in both in vitro and cell-based assays. These findings endorse further optimization efforts based on this structural phenotype to develop potential anticancer drug candidates.
Co-reporter:Cheng Fang, Zhiyan Xiao, Zongru Guo
Journal of Molecular Graphics and Modelling 2011 Volume 29(Issue 6) pp:800-808
Publication Date(Web):April 2011
DOI:10.1016/j.jmgm.2011.01.003
A three-dimensional (3D) pharmacophore modelling approach was applied to a diverse data set of known cyclin-dependent kinase 9 (CDK9) inhibitors. Diversity sampling and principal components analysis (PCA) were employed to ensure the rational selection of representative training sets. Twelve statistically robust pharmacophore models were generated using the HypoGen algorithm. The resulting models showed high homology and indicated great convergence in ascertaining pharmacophoric features essential for CDK9 inhibitory activity. One of the best models (Hypo 6) was assessed further by external predictive capability, randomization test, as well as its performance in virtual screening. The capability of the resulting models to reliably predict the inhibitory activity of external data sets and discriminate active structures from general databases would assist the identification and optimization of novel CDK9 inhibitors.Graphical abstractResearch highlights► 75 diverse known CDK9 inhibitors were used for model generation. ► Representative training sets were selected by diversity sampling. ► Predictive pharmacophore models were generated with HypoGen algorithm. ► The model was validated with multiple approaches. ► The model showed good predictive capability and discriminative power.
Co-reporter:Wen Li Xi, Qian Cai, Yan Bo Tang, Hua Sun, Zhi Yan Xiao
Chinese Chemical Letters 2010 Volume 21(Issue 10) pp:1153-1156
Publication Date(Web):October 2010
DOI:10.1016/j.cclet.2010.03.040
In order to investigate the effect of different C4 linkage moieties on the cytotoxicity of podophyllotoxin derivatives, novel 4-N- and 4-C-substituted 4′-O-demethylepipodophyllotoxin derivatives were designed and synthesized. All the compounds were tested against A549 and MCF-7 tumor cells in vitro, and six compounds showed significant cytotoxicity. The most active compound 9f was superior to GL-331, and exhibited potent cytotoxicity with IC50 value at 10−7 mol/L level.

![2H-Indol-2-one, 3-[(3E)-1,3-dihydro-3-(hydroxyimino)-2H-indol-2-ylidene]-1,3-dihydro-, (3Z)-](/data/chemimg/127100/667463-82-3.png)
![2H-Indol-2-one, 3-[(3E)-1,3-dihydro-3-(hydroxyimino)-2H-indol-2-ylidene]-1,3-dihydro-, (3Z)-](/data/chemimg/127100/667463-82-3_b.png)
![(1R,7S,8R,9R)-9-[(1E,3E)-4-CARBOXY-1,3-PENTADIEN-1-YL]-7-HYDROXY-9-METHYL-11-OXO-10-OXATRICYCLO[6.3.2.0<SUP>1,7</SUP>]TRIDEC-3-ENE-4-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/383900/383870-96-0.png)
![(1R,7S,8R,9R)-9-[(1E,3E)-4-CARBOXY-1,3-PENTADIEN-1-YL]-7-HYDROXY-9-METHYL-11-OXO-10-OXATRICYCLO[6.3.2.0<SUP>1,7</SUP>]TRIDEC-3-ENE-4-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/383900/383870-96-0_b.png)


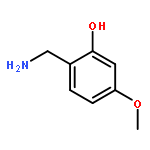
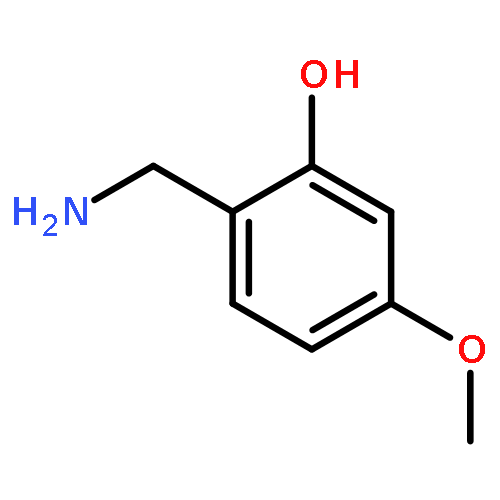
![L-Valine, N-[N6-[(phenylmethoxy)carbonyl]-L-lysyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/150100/150035-95-3.png)
![L-Valine, N-[N6-[(phenylmethoxy)carbonyl]-L-lysyl]-, phenylmethyl ester](http://img.cochemist.com/ccimg/150100/150035-95-3_b.png)



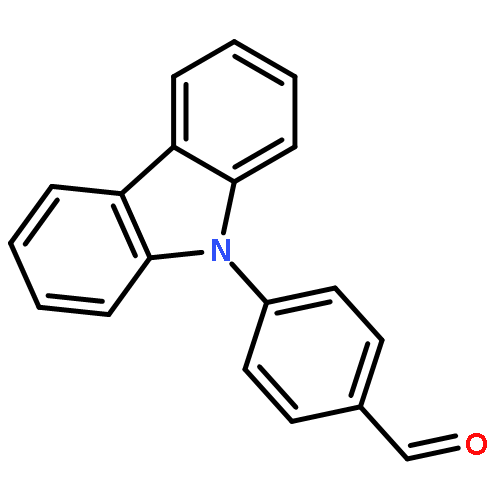
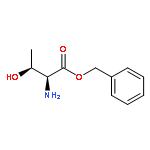
![Ethanone, 1-[3,5-bis(1,1-dimethylethyl)-4-methoxyphenyl]-](http://img.cochemist.com/ccimg/30500/30492-50-3.png)
![Ethanone, 1-[3,5-bis(1,1-dimethylethyl)-4-methoxyphenyl]-](http://img.cochemist.com/ccimg/30500/30492-50-3_b.png)





