Co-reporter:Jia Wei, Leila M. Duman, Daniel W. Redman, Brendan L. Yonke, Peter Y. Zavalij, and Lawrence R. Sita
Organometallics November 13, 2017 Volume 36(Issue 21) pp:4202-4202
Publication Date(Web):October 19, 2017
DOI:10.1021/acs.organomet.7b00643
A new structurally diverse ligand for metal complexes, the (κ2-N,N)-iminocaprolactam group, that can be obtained in large bulk quantities from inexpensive and readily available starting materials, is introduced. In the present report, the ability of this family of ligands to provide a viable substitute for the acyclic amidinate moiety of a class of cationic monocyclopentadienyl, amidinate (CPAM) group 4 metal initiators for the living coordination polymerization (LCP) and living coordinative chain transfer polymerization (LCCTP) of α-olefins is documented through the synthesis of (η5-C5Me5)Hf[(R)NimcapN](Me)2 [R = benzyl (1), CH2(1-naphthyl) (2), and tert-butyl [C(CH3)3] (3)] as preinitiators for the (stereoselective) LCP and LCCTP of propene.
Co-reporter:Samantha R. Nowak, Wonseok Hwang, and Lawrence R. Sita
Journal of the American Chemical Society April 19, 2017 Volume 139(Issue 15) pp:5281-5281
Publication Date(Web):April 3, 2017
DOI:10.1021/jacs.6b13285
Spin-casting of a cellobiose-atactic polypropene (CB-aPP) conjugate (1) from a 0.1% (w/w) n-butanol/hexane solution onto highly oriented pyrolytic graphite (HOPG) and carbon-coated Si(100) spontaneously produced microphase-separated sub-10-nm nanostructured ultrathin films in the form of alternating CB and aPP lamellar domains (d = 6.60 ± 0.68 nm) that are oriented perpendicular to the substrate surface. Thermal annealing at modest temperatures (e.g., 50–100 °C), and as low as the physiologically relevant temperature of 38 °C, serves to drive a structural transition that yields a parallel stacked bilayer assembly as the thermodynamically favored nanostructure. These results establish the advantage of low molecular weight, narrow polydispersity, and amorphous, low Tg, poly(α-olefinate)s (xPAOs) as a new class of hydrophobic building block for amphiphilic materials, and sugar–PAO conjugates in particular, for the development of stimuli-responsive, nanostructured materials for technological applications at physiological temperatures.
Co-reporter:Leila M. Duman, Wesley S. Farrell, Peter Y. Zavalij, and Lawrence R. Sita
Journal of the American Chemical Society 2016 Volume 138(Issue 45) pp:14856-14859
Publication Date(Web):October 25, 2016
DOI:10.1021/jacs.6b09789
Programmed manipulation of the subtle interplay of nonbonded steric interactions within a supporting ligand environment has been used for the conversion of a photochemically driven chemical cycle for group 6 metal-mediated nitrogen fixation into a thermally promoted process with increased energy efficiency and atom economy for key transformations involving N≡N bond cleavage and N-atom functionalization of coordinated N2.
Co-reporter:Wesley S. Farrell, Brendan L. Yonke, Jonathan P. Reeds, Peter Y. Zavalij, and Lawrence R. Sita
Organometallics 2016 Volume 35(Issue 8) pp:1132-1140
Publication Date(Web):April 14, 2016
DOI:10.1021/acs.organomet.6b00131
A series of midvalent monocyclopentadienyl monoamidinate (CPAM) group 6 complexes of the general formula Cp*[N(iPr)C(Me)N(iPr)]M(CO)(L) (II), where Cp* = η5-C5Me5 and M = Mo, W, have been prepared, and most of them have been structurally characterized. Treatment of the ditungsten “end-on-bridged” dinitrogen complex {Cp*[N(iPr)C(Me)N(iPr)]W}2(μ-η1:η1-N2) (3) with excess NCMe under a CO atmosphere produced the ditungsten bridging diimido complex {Cp*[N(iPr)C(Me)N(iPr)]W}2[μ-η1:η1-NC(Me)═C(Me)N] (4). Photolysis of Cp*[N(iPr)C(Me)N(iPr)]M(CO)2, where M = Mo (6), W (7), or treatment of Cp*[N(iPr)C(Me)N(iPr)]Mo(CO)(NCMe) (1a) with excess alkene provided Cp*[N(iPr)C(Me)N(iPr)]M(CO)(L) for M = Mo and L = η2-ethene (8), M = W and L = η2-ethene (9), M = Mo and L = η2-norbornene (10), M = W and L = η2-norbornene (11), M = W and L = η2-cyclopentene (12), M = Mo and L = η2-cyclopentene (13), and M = Mo and L = η2-styrene (14). When isobutene was employed as the alkene, C–H bond activation occurred to produce Cp*[N(iPr)C(Me)N(iPr)]W(H)(η3-C4H7) (15). Photolysis of 7 in the presence of SMe2 provided Cp*[N(iPr)C(Me)N(iPr)]W[κ-C,O-C(O)Me](SMe) (16) through oxidative C–S bond activation of a coordinated SMe2, followed by 1,1-carbonyl migratory insertion into the new W–C bond. Finally, reaction of 1a with propylene oxide (C3H6O) provided the 16-electron complex Cp*[N(iPr)C(Me)N(iPr)]Mo[C(O)CH(Me)CH2O] (19) via similar oxidative C–O bond activation of coordinated C3H6O, followed by 1,1-carbonyl migratory insertion into the Mo–C bond of an intermediate metallaoxetane. Under a CO atmosphere, 19 is converted to the 18-electron complex Cp*[N(iPr)C(Me)N(iPr)]Mo[C(O)CH(Me)CH2O](CO) (20). These results provide additional support for the development of new stoichiometric and catalytic transformations that are mediated by CPAM group 6 metal complexes and that are relevant to the goal of small-molecule fixation.
Co-reporter:Tessy S. Thomas;Wonseok Hwang ; Lawrence R. Sita
Angewandte Chemie International Edition 2016 Volume 55( Issue 15) pp:4683-4687
Publication Date(Web):
DOI:10.1002/anie.201600035
Abstract
Living coordinative chain-transfer polymerization of α-olefins, followed by chemical functionalization of a Zn(polymeryl)2 intermediate, provides entry to end-group functionalized poly(α-olefinates) (x-PAOs) that can serve as a new class of non-polar building block with tailorable occupied volumes. Application of these x-PAOs for the synthesis and self-assembly of sugar-polyolefin hybrid conjugates demonstrate the ability to manipulate the morphology of the ultra-thin film nanostructure through variation in occupied volume of the x-PAO domain.
Co-reporter:Tessy S. Thomas;Wonseok Hwang ; Lawrence R. Sita
Angewandte Chemie 2016 Volume 128( Issue 15) pp:4761-4765
Publication Date(Web):
DOI:10.1002/ange.201600035
Abstract
Living coordinative chain-transfer polymerization of α-olefins, followed by chemical functionalization of a Zn(polymeryl)2 intermediate, provides entry to end-group functionalized poly(α-olefinates) (x-PAOs) that can serve as a new class of non-polar building block with tailorable occupied volumes. Application of these x-PAOs for the synthesis and self-assembly of sugar-polyolefin hybrid conjugates demonstrate the ability to manipulate the morphology of the ultra-thin film nanostructure through variation in occupied volume of the x-PAO domain.
Co-reporter:Wesley S. Farrell, Peter Y. Zavalij, and Lawrence R. Sita
Organometallics 2016 Volume 35(Issue 14) pp:2361-2366
Publication Date(Web):July 1, 2016
DOI:10.1021/acs.organomet.6b00302
In the presence of excess amounts of elemental sulfur, the dimolybdenum dinitrogen complex {Cp*Mo[N(iPr)C(Ph)N(iPr)]}2(μ-N2) (4; Cp* = η5-C5Me5) serves as a precatalyst for the production of isothiocyanates from isonitriles via highly efficient and atom-economical metal-mediated sulfur atom transfer (SAT) under mild conditions. Mechanistic and structural studies support a catalytic cycle for SAT involving initial formation of a Mo(II) bis(isonitrile) complex that then undergoes sulfination to generate a formal “side-bound” Mo(IV) κ2-(C,S)-isothiocyanate as the key intermediate. This metal-catalyzed SAT process has further been employed for the “on-demand” production of isothiocyanates that are trapped in situ by benzhydrazides to provide thiosemicarbazides, which are useful precursors to biologically active thiadiazoles.
Co-reporter:Kaitlyn E. Crawford and Lawrence R. Sita
ACS Macro Letters 2015 Volume 4(Issue 9) pp:921
Publication Date(Web):August 14, 2015
DOI:10.1021/acsmacrolett.5b00447
Sequential cyclic/linear/cyclic living coordination polymerization of 1,6-heptadiene (HPD), propene, and HPD, respectively, employing the well-defined and soluble group 4 transition-metal initiator, {(η5-C5Me5)Hf(Me)[N(Et)C(Me)N(Et)]}[B(C6F5)4], provides the stereoirregular, amorphous poly(1,3-methylenecyclohexane)-b-atactic polypropene-b-poly(1,3-methylenecyclohexane) (PMCH-b-aPP-b-PMCH) polyolefin triblock copolymer (I) in excellent yield. By varying the weight fraction of the end group, minor component “hard” PMCH block domains, fPMCH, relative to that of the midblock “soft” aPP domain, three different compositional grades of these polyolefin block copolymers, Ia–c, were prepared and shown by AFM and TEM to adopt microphase-separated morphologies in the solid state, with spherical and cylindrical morphologies being observed for fPMCH = 0.09 (Ia) and 0.23 (Ic), respectively, and a third more complex morphology being observed for Ib (fPMCH = 0.17). Tensile testing of Ia–c served to establish these materials as a new structural class of polyolefin thermoplastic elastomers, with Ia being associated with superior elastic recovery (94 ± 1%) after each of several stress–strain cycles.
Co-reporter:Andrew J. Keane;Wesley S. Farrell;Brendan L. Yonke;Peter Y. Zavalij ; Lawrence R. Sita
Angewandte Chemie International Edition 2015 Volume 54( Issue 35) pp:10220-10224
Publication Date(Web):
DOI:10.1002/anie.201502293
Abstract
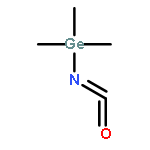
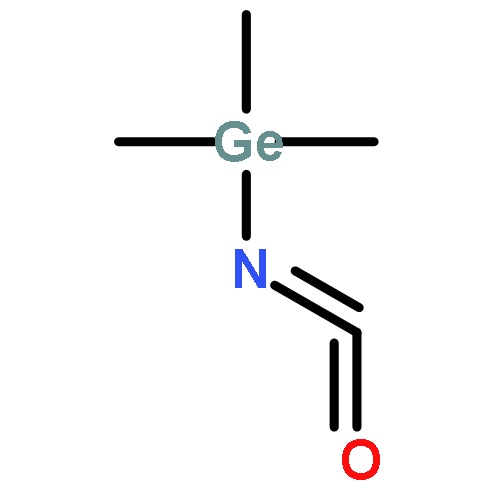
A highly efficient and versatile chemical cycle has been developed for the production of isocyanates through the molecular fixation of N2, CO2 and R3ECl (E=C, Si, and Ge). Key steps include a ‘one-pot’ photolytic NN bond cleavage of a Group 6 dinuclear dinitrogen complex with in situ trapping by R3ECl to provide a metal terminal imido complex that can engage in simultaneous nitrene-group transfer and oxygen-atom transfer to generate an intermediate metal terminal oxo complex with release of the isocyanate product. Reaction of the oxo complex with additional equivalents of R3ECl regenerates a metal dichloride that is the precursor for dinuclear dinitrogen starting material.
Co-reporter:Andrew J. Keane;Wesley S. Farrell;Brendan L. Yonke;Peter Y. Zavalij ; Lawrence R. Sita
Angewandte Chemie 2015 Volume 127( Issue 35) pp:10358-10362
Publication Date(Web):
DOI:10.1002/ange.201502293
Abstract
A highly efficient and versatile chemical cycle has been developed for the production of isocyanates through the molecular fixation of N2, CO2 and R3ECl (E=C, Si, and Ge). Key steps include a ‘one-pot’ photolytic NN bond cleavage of a Group 6 dinuclear dinitrogen complex with in situ trapping by R3ECl to provide a metal terminal imido complex that can engage in simultaneous nitrene-group transfer and oxygen-atom transfer to generate an intermediate metal terminal oxo complex with release of the isocyanate product. Reaction of the oxo complex with additional equivalents of R3ECl regenerates a metal dichloride that is the precursor for dinuclear dinitrogen starting material.
Co-reporter:Wesley S. Farrell;Peter Y. Zavalij ; Lawrence R. Sita
Angewandte Chemie 2015 Volume 127( Issue 14) pp:4343-4347
Publication Date(Web):
DOI:10.1002/ange.201410353
Abstract
The group 6 molybdenum(II) cyclopentadienyl amidinate (CPAM) bis(carbonyl) complex [Cp*Mo{N(iPr)C(Ph)N(iPr)}(CO)2] (Cp*=η5-C5Me5) serves as a precatalyst for the high-yielding photocatalytic production of COS from CO and S8 under near-ambient conditions (e.g., 10 psi, 25 °C). Further documented is the isolation and structural characterization of several key transition-metal intermediates which collectively support a novel molybdenum(IV)-based catalytic cycle as being operative. Finally, in the presence of an excess amount of a primary amine, it is demonstrated that this catalytic system can be successfully used for the “on-demand” generation and utilization of COS as a chemical reagent for the synthesis of ureas.
Co-reporter:Wesley S. Farrell;Peter Y. Zavalij ; Lawrence R. Sita
Angewandte Chemie International Edition 2015 Volume 54( Issue 14) pp:4269-4273
Publication Date(Web):
DOI:10.1002/anie.201410353
Abstract
The group 6 molybdenum(II) cyclopentadienyl amidinate (CPAM) bis(carbonyl) complex [Cp*Mo{N(iPr)C(Ph)N(iPr)}(CO)2] (Cp*=η5-C5Me5) serves as a precatalyst for the high-yielding photocatalytic production of COS from CO and S8 under near-ambient conditions (e.g., 10 psi, 25 °C). Further documented is the isolation and structural characterization of several key transition-metal intermediates which collectively support a novel molybdenum(IV)-based catalytic cycle as being operative. Finally, in the presence of an excess amount of a primary amine, it is demonstrated that this catalytic system can be successfully used for the “on-demand” generation and utilization of COS as a chemical reagent for the synthesis of ureas.
Co-reporter:Andrew J. Keane ; Brendan L. Yonke ; Masakazu Hirotsu ; Peter Y. Zavalij
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9906-9909
Publication Date(Web):June 24, 2014
DOI:10.1021/ja505309j
Experimental data support a mechanism for N≡N bond cleavage within a series of group 5 bimetallic dinitrogen complexes of general formula, {Cp*M[N(iPr)C(R)N(iPr)]}2(μ-N2) (Cp* = η5-C5Me5) (M = Nb, Ta), that proceeds in solution through an intramolecular “end-on-bridged” (μ-η1:η1-N2) to “side-on-bridged” (μ-η2:η2-N2) isomerization process to quantitatively provide the corresponding bimetallic bis(μ-nitrido) complexes, {Cp*M[N(iPr)C(R)N(iPr)](μ-N)}2. It is further demonstrated that subtle changes in the steric and electronic features of the distal R-substituent, where R = Me, Ph and NMe2, can serve to modulate the magnitude of the free energy barrier height for N≡N bond cleavage as assessed by kinetic studies and experimentally derived activation parameters. The origin of the contrasting kinetic stability of the first-row congener, {Cp*V[N(iPr)C(Me)N(iPr)]}2(μ-η1:η1-N2) toward N≡N bond cleavage is rationalized in terms of a ground-state electronic structure that favors a significantly less-reduced μ-N2 fragment.
Co-reporter:Kaitlyn E. Crawford and Lawrence R. Sita
ACS Macro Letters 2014 Volume 3(Issue 6) pp:506
Publication Date(Web):May 13, 2014
DOI:10.1021/mz500126r
Transition-metal-mediated coordination cyclopolymerization of bis(2-propenyl)dimethylsilane (1a) using the C1-symmetric, group 4 metal preinitiator, (η5-C5Me5)Zr(Me)2[N(Et)C(Me)N(tBu)] (I), in combination with 1 equiv of the borate coinitiator, [PhNHMe2][B(C6F5)4] (II), proceeds in a regio- and stereospecific manner to provide highly stereoregular 3,5-cis,isotactic poly(3,5-methylene-1,1-dimethyl-1-silacyclohexane) (2a). Successful stereoengineering of 2a to eliminate undesirable crystallinity while preserving a high Tg value of >120 °C was subsequently accomplished by employing a “two-state” propagation system that uniquely produces an isotactic stereoblock microstructure of decreasing stereoblock length with decreasing percent level of “activation” of I with II. The controlled character of cyclopolymerization of 1a using the less sterically encumbered preinitiator, (η5-C5Me5)Hf(Me)2[N(Et)C(Me)N(Et)] (III), and 1 equiv of II was used to prepare well-defined poly(1-hexene)-b-poly(3,5-methylene-1-silacyclohexane) block copolymers through sequential monomer additions.
Co-reporter:Brendan L. Yonke, Jonathan P. Reeds, Philip P. Fontaine, Peter Y. Zavalij, and Lawrence R. Sita
Organometallics 2014 Volume 33(Issue 13) pp:3239-3242
Publication Date(Web):June 26, 2014
DOI:10.1021/om500532s
Under an atmosphere of CO, the Mo(IV) imido complex Cp*Mo[N(iPr)C(Me)N(iPr)](NSiMe3) (Cp* = η5-C5Me5) (1) serves as a catalyst for production of an isocyanate via metal-mediated nitrene group transfer in benzene solution under mild conditions (55 °C, 10 psi) according to RN3 + CO → N2 + RNCO. Mechanistic and structural studies support a catalytic cycle for nitrene group transfer involving formal Mo(II) monocarbonyl and Mo(IV) (κ2-C,N)-isocyanate intermediates. These results complement an earlier finding that catalytic production of isocyanates can alternatively proceed through oxygen-atom transfer and an isomeric Mo(IV) (κ2-C,O)-isocyanate according to N2O + CNR → N2 + RNCO.
Co-reporter:Kaitlyn E. Crawford
Journal of the American Chemical Society 2013 Volume 135(Issue 24) pp:8778-8781
Publication Date(Web):May 22, 2013
DOI:10.1021/ja402262x
External control over the rate of dynamic methyl group exchange between configurationally stable active species and configurationally unstable dormant species with respect to chain-growth propagation within a highly stereoselective and regiospecific living coordination polymerization of 1,6-heptadiene has been used to generate a spectrum of different physical forms of poly(1,3-methylenecyclohexane) (PMCH) in which the stereochemical microstructure has been systematically varied between two limiting forms. The application of this strategy to manipulate the bulk properties of PMCH and the solid-state microphase behavior of well-defined PMCH-b-poly(1-hexene) block copolymers is further demonstrated.
Co-reporter:Andrew J. Keane ; Peter Y. Zavalij
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9580-9583
Publication Date(Web):May 31, 2013
DOI:10.1021/ja402804k
Chemical reduction of the Ta(V) hydrazido chloride 1 generates the open-shell, mononuclear Ta(IV) hydrazido complex 2, which upon N-methylation yields the corresponding structurally characterized Ta(IV) hydrazidium 6. Chemical reduction of 6 results in N–N bond cleavage to generate a cis/trans mixture of the [Ta(V),Ta(V)] bis(μ-nitrido) product 7 in tetrahydrofuran and the mononuclear Ta(V) parent imide 8 in toluene. These results serve to establish an important foundation for the pursuit of a group-5 metal variant of the Schrock cycle for dinitrogen fixation.
Co-reporter:Jia Wei ; Wonseok Hwang ; Wei Zhang
Journal of the American Chemical Society 2013 Volume 135(Issue 6) pp:2132-2135
Publication Date(Web):January 23, 2013
DOI:10.1021/ja312463f
Modulation of steric interactions remote from the active sites within a series of dinuclear bis-propagators derived from racemic 2–4 was used to attenuate the rate of reversible chain transfer between active transition-metal centers and excess equivalents of inactive main-group-metal alkyl species relative to chain growth propagation, as a strategy for achieving the stereoselective living coordinative chain transfer polymerization of propene to provide isotactic stereoblock polypropene. Under identical conditions, the corresponding mononuclear propagator derived from racemic 1 produced only atactic polypropene.
Co-reporter:Brendan L. Yonke, Andrew J. Keane, Peter Y. Zavalij, and Lawrence R. Sita
Organometallics 2012 Volume 31(Issue 1) pp:345-355
Publication Date(Web):December 19, 2011
DOI:10.1021/om200961r
Two independent synthetic routes to the monocyclopentadienyl, amidinate (CpAm) and guanidinate (CpGu) mononuclear Ta(IV, d1) terminal imido complexes Cp*Ta[N(iPr)C(X)N(iPr)][N(tBu)] (Cp* = η5-C5Me5) for X = Me (1) and NMe2 (2), respectively, were developed. For 1, synthesis proceeded via the amido, chloride intermediate Cp*Ta[N(iPr)C(Me)N(iPr)](Cl)[NH(tBu)] (4), which was kinetically deprotonated with LiN(iPr)2 to yield the enamido, amido species Cp*Ta[N(iPr)C(CH2)N(iPr)[NH(tBu)] (5). In toluene solution, 5 underwent quantitative tautomerization to the desired CpAm terminal imido 1. For 2, the amido, chloride intermediate Cp*Ta[N(iPr)C(NMe2)N(iPr)](Cl)[NH(tBu)] (8) was first synthesized and then reacted with TEMPO to provide the Ta(V) imido chloride Cp*Ta[N(iPr)C(NMe2)N(iPr)](Cl)[N(tBu)] (9) through oxidative hydrogen-atom abstraction of the amido group. Chemical reduction of 9 with potassium graphite (KC8) then served to provide the desired CpGu terminal imido 2. All new compounds were structurally characterized by single-crystal X-ray analysis. Although 1 and 2 proved to be unreactive toward hydrogenation or hydrosilyation involving the Ta═N bond, 2 was shown to engage in radical-based chemistry with MeI and PhS-SPh to yield the Ta(V) imido complexes Cp*Ta[N(iPr)C(NMe2)N(iPr)](X)[N(tBu)], where X = Me (10), I (11), and SPh (12). A similar radical-based reaction of {Cp*Ta[N(iPr)C(Me)N(iPr)]}2(μ-η1:η1-N2) (I) with PhS-SPh yielded the product of formal 1,4-addition, {Cp*Ta[N(iPr)C(Me)N(iPr)](SPh)}2(μ-η1:η1-N2) (13).
Co-reporter:Jonathan P. Reeds ; Brendan L. Yonke ; Peter Y. Zavalij
Journal of the American Chemical Society 2011 Volume 133(Issue 46) pp:18602-18605
Publication Date(Web):October 25, 2011
DOI:10.1021/ja208669s
In the presence of CO, facile N–N bond cleavage of N2O occurs at the formal Mo(II) center within coordinatively unsaturated mononuclear species derived from Cp*Mo[N(iPr)C(Me)N(iPr)](CO)2 (Cp* = η5-C5Me5) (1) and {Cp*Mo[N(iPr)C(Me)N(iPr)]}2(μ-η1:η1-N2) (9) under photolytic and dark conditions, respectively, to produce the nitrosyl, isocyanate complex Cp*Mo[N(iPr)C(Me)N(iPr)](κ-N-NO)(κ-N-NCO) (7). Competitive N–O bond cleavage of N2O proceeds under the same conditions to yield the Mo(IV) terminal metal oxo complex Cp*Mo[N(iPr)C(Me)N(iPr)](O) (3), which can be recycled to produce more 7 through oxygen-atom-transfer oxidation of CO to produce CO2.
Co-reporter:Brendan L. Yonke;Jonathan P. Reeds;Peter Y. Zavalij ; Lawrence R. Sita
Angewandte Chemie International Edition 2011 Volume 50( Issue 51) pp:12342-12346
Publication Date(Web):
DOI:10.1002/anie.201106074
Co-reporter:Brendan L. Yonke;Jonathan P. Reeds;Peter Y. Zavalij ; Lawrence R. Sita
Angewandte Chemie 2011 Volume 123( Issue 51) pp:12550-12554
Publication Date(Web):
DOI:10.1002/ange.201106074
Co-reporter: Lawrence R. Sita
Angewandte Chemie International Edition 2011 Volume 50( Issue 31) pp:6963-6965
Publication Date(Web):
DOI:10.1002/anie.201101913
Co-reporter: Lawrence R. Sita
Angewandte Chemie 2011 Volume 123( Issue 31) pp:7097-7099
Publication Date(Web):
DOI:10.1002/ange.201101913
Co-reporter:Philip P. Fontaine ; Brendan L. Yonke ; Peter Y. Zavalij
Journal of the American Chemical Society 2010 Volume 132(Issue 35) pp:12273-12285
Publication Date(Web):August 13, 2010
DOI:10.1021/ja100469f
Chemical reduction of Cp*M[N(i-Pr)C(Me)N(i-Pr)]Cl3 (Cp* = η5-C5Me5) (1, M = Mo) and (2, M = W) using 0.5% NaHg in THF provided excellent yields of the diamagnetic dinuclear end-on-bridged dinitrogen complexes {Cp*M[N(i-Pr)C(Me)N(i-Pr)]}2(μ-η1:η1-N2) (6, M = Mo) and (8, M = W), respectively. Chemical reduction of Cp*Mo[N(i-Pr)C(NMe2)N(i-Pr)]Cl2 (4) with 3 equiv of KC8 in THF similarly yielded diamagnetic {Cp*Mo[N(i-Pr)C(NMe2)N(i-Pr)]}2(μ-η1:η1-N2) (7). Single-crystal X-ray analyses of 7 and 8 confirmed the dinuclear end-on-bridged μ-η1:η1-N2 coordination mode and the solid-state molecular structures of these compounds provided d(NN) values of 1.267(2) and 1.277(8) Å for 7 and 8, respectively. Based on a comparison of 15N NMR spectra for 15N2 (99%)-labeled 6 and 15N2 (99%)-labeled 8, as well as similarities in chemical reactivity, a dinuclear μ-η1:η1-N2 structure for 6 is further proposed. For comparison with a first-row metal derivative, chemical reduction of Cp*Ti[N(i-Pr)C(Me)N(i-Pr)]Cl2 (9) with KC8 in THF was conducted to provide {Cp*Ti[N(i-Pr)C(Me)N(i-Pr)]}2(μ-η1:η1-N2) (10) for which a d(NN) value of 1.270(2) Å was obtained through X-ray crystallography. Compounds 6−8 were all found to be thermally robust in toluene solution up to temperatures of at least 100 °C, and 6 and 8 were determined to be inert toward the addition of H2 or H3SiPh under a variety of conditions. Single-crystal X-ray analysis of meso-{Cp*Mo(H)[N(i-Pr)C(Me)N(i-Pr)]}2(μ-η1:η1-N2) (meso-11), which was serendipitously isolated as a product of attempted alkylation of Cp*Mo[N(i-Pr)C(Me)N(i-Pr)]Cl2 (3) with 2 equiv of n-butyllithium, revealed a smaller d(NN) value of 1.189(4) Å that is consistent with two Mo(IV,d2) centers connected by a bridging diazenido, [μ-N2]2−, moiety. Moreover, meso-11 was found to undergo clean dehydrogenation in solution at 50 °C to provide 6 via a first-order process. Chemical oxidation of 8 with an excess of PbCl2 in toluene solution at 25 °C provided a 1:1 mixture of rac- and meso-{Cp*W(Cl)[N(i-Pr)C(Me)N(i-Pr)]}2(μ-η1:η1-N2) (12); both isomers of which provided solid-state structures through X-ray analyses that are consistent with an electronic configuration comprised of two W(IV,d2) centers linked through a bridging [N2]2− group [cf. for rac-12, d(NN) = 1.206(9) Å, and for meso-12, d(NN) = 1.192(3) Å]. Finally, treatment of 6 and 8 with either 4 equiv of CNAr (Ar = 3,5-Me2C6H3) or an excess of CO in toluene provided excellent yields of Cp*M[N(i-Pr)C(Me)N(i-Pr)](CNAr)2 (13, M = Mo and 14, M = W) and Cp*M[N(i-Pr)C(Me)N(i-Pr)](CO)2 (15, M = Mo and 16, M = W), respectively. Single-crystal X-ray analyses of 13−16, along with observation of reduced IR vibrational νCN or νCO bond-stretching frequencies, provide strong support for the electron-rich character of the Cp*M[N(i-Pr)C(Me)N(i-Pr)] fragment that can engage in a high degree of back-donation with moderate to strong π-acceptors, such as N2, CNR, and CO. The collective results of this work are analyzed in terms of the possible steric and electronic factors that contribute to preferred mode of μ-N2 coordination and the extent of N≡N activation, including complete N−N bond scission, within the now completed experimentally-derived ligand-centered isostructural series of {Cp*M[N(i-Pr)C(Me)N(i-Pr)]}2(μ-N2) compounds where M = Ti, Zr, Hf, Ta, Mo, and W.
Co-reporter:Jia Wei;Wei Zhang ;LawrenceR. Sita
Angewandte Chemie 2010 Volume 122( Issue 10) pp:1812-1816
Publication Date(Web):
DOI:10.1002/ange.200906658
Co-reporter:Jia Wei;Wei Zhang;Rennisha Wickham ; Lawrence R. Sita
Angewandte Chemie 2010 Volume 122( Issue 48) pp:9326-9330
Publication Date(Web):
DOI:10.1002/ange.201004709
Co-reporter:Jia Wei;Wei Zhang ;LawrenceR. Sita
Angewandte Chemie International Edition 2010 Volume 49( Issue 10) pp:1768-1772
Publication Date(Web):
DOI:10.1002/anie.200906658
Co-reporter:Jia Wei;Wei Zhang;Rennisha Wickham ; Lawrence R. Sita
Angewandte Chemie International Edition 2010 Volume 49( Issue 48) pp:9140-9144
Publication Date(Web):
DOI:10.1002/anie.201004709
Co-reporter:Emily F. Trunkely, Albert Epshteyn, Peter Y. Zavalij, and Lawrence R. Sita
Organometallics 2010 Volume 29(Issue 23) pp:6587-6593
Publication Date(Web):November 12, 2010
DOI:10.1021/om100877d
Alkylation of Cp*TiCl2[N(i-Pr)C(Me)N(i-Pr)] (Cp* = η5-C5Me5) (1) with two equivalents of an alkyllithium reagent provided high yields of the thermally stable, crystalline, paramagnetic titanium(III) alkyl complexes of general structure Cp*Ti(R)[N(i-Pr)C(Me)N(i-Pr)] where R = Et (2), n-Bu (3), i-Bu (4), neopentyl (5), and n-hexyl (6). Solid-state structural characterization of compounds 2−5 by single-crystal X-ray analysis revealed the absence of α- or β-hydrogen agostic interactions between the metal center and the alkyl group R. Isocyanides (R′NC) undergo quantitative 1,1-insertion into the titanium−carbon bond of the alkyl group of 2−6 to provide high yields of the corresponding series of crystalline, paramagnetic Ti(III) η2-iminoacyl derivatives, Cp*Ti[η2-N(R′)═CR)][N(i-Pr)C(Me)N(i-Pr)] (7−11, respectively), which were also structurally characterized by X-ray crystallography for compounds 7 and 9−11. Finally, compounds 3−5 were oxidized with PbCl2 in diethyl ether to cleanly generate the respective Ti(IV) monochloro, monoalkyl complexes Cp*Ti(R)(Cl)[N(i-Pr)C(Me)N(i-Pr)] (12−14, respectively), the solid-state structures of which were also determined by single-crystal X-ray analyses.
Co-reporter:LawrenceR. Sita
Angewandte Chemie International Edition 2009 Volume 48( Issue 14) pp:2464-2472
Publication Date(Web):
DOI:10.1002/anie.200802661
Co-reporter:LawrenceR. Sita
Angewandte Chemie 2009 Volume 121( Issue 14) pp:2500-2508
Publication Date(Web):
DOI:10.1002/ange.200802661
Co-reporter:Albert Epshteyn, Emily F. Trunkely, Denis A. Kissounko, James C. Fettinger and Lawrence R. Sita
Organometallics 2009 Volume 28(Issue 8) pp:2520-2526
Publication Date(Web):March 27, 2009
DOI:10.1021/om900132u
Ring-expansion of the zirconacyclopropane (η5-C5Me5)Zr[N(i-Pr)C(Me)N(i-Pr)](η2-CH2CHC6H5) (1) was accomplished through insertion of ethene, propene, and styrene into a metal−carbon bond to yield the series of crystalline zirconacyclopentane derivatives 2−4, respectively, for which solid-state molecular structures have been obtained through single-crystal X-ray analyses. The metallacyclopentane rings of 2 and 4 adopt half-chair conformations that place all β-hydrogens distant from the metal center (cf. nonbonded distances of 3.2−4.0 Å). On the other hand, a twisted-envelope conformation for the zirconacyclopentane ring of 3 features a β-hydrogen agostic interaction with the metal center [cf. a Zr1−H21B distance of 2.38(2) Å]. Unlike 2 and 4, compound 3 decomposes rapidly in solution at 25 °C to provide the structurally characterized zirconacyclopent-3-ene derivative 5. Ring-expansion of 1 with 1-(trimethylsilyl)ethene directly provides the zirconacyclopent-3-ene derivative 7. A mechanism for this facile metallacyclopentane dehydrogenative process is proposed that features an intramolecular allylic β-hydrogen abstraction within a 3-butenyl metal hydride intermediate as a key step. It is further proposed that similar metallacyclopentane to metallacyclopent-3-ene dehydrogenations may be more common than previously thought.
Co-reporter:Wei Zhang ;LawrenceR. Sita
Advanced Synthesis & Catalysis 2008 Volume 350( Issue 3) pp:439-447
Publication Date(Web):
DOI:10.1002/adsc.200700506
Abstract
The series of bimetallic complexes, [(η5-C5Me5)Zr(Me)2]2 [N(t-Bu)C(Me)N(CH2)nNC(Me)N(t-Bu)] 3 (n=8), 4 (n=6), and 5 (n=4) were prepared in high yield through a simple, one-pot synthesis involving 2 equiv. of in situ generated (η5-C5Me5)Zr(Me)3 and the corresponding bis-carbodiimide, (t-Bu)NCN(CH2)nNCN(t-Bu). Compounds 3–5 were found to be highly isoselective for the living Ziegler–Natta polymerization of propene upon 100% activation using 2 equiv. of the borate co-initiator, [PhNHMe2] [B(C6F5)4] (2), with the degree of stereoselectivity decreasing slightly as the two metal centers are brought closer together [cf., 3 (σ=0.92)>4 (σ=0.91)>5 (σ=0.89)]. Under conditions of sub-stoichiometric activation by 2, all three bimetallic initiators, 3–5, were found to engage in degenerative transfer living Ziegler–Natta polymerization involving rapid and reversible methyl group transfer between active, (cationic) and dormant, (neutral) methyl, polymeryl zirconium centers. Under these conditions, the frequency of mr triad stereoerror incorporation into the polypropene (PP) microstructure decreases as the two metal centers are brought closer together as a result of increasing barriers for metal-centered epimerization within the neutral metal site due to correspondingly greater non-bonded steric interactions vis-à-vis mononuclear 1.
Co-reporter:Wei Zhang, Jia Wei and Lawrence R. Sita
Macromolecules 2008 Volume 41(Issue 21) pp:7829-7833
Publication Date(Web):October 15, 2008
DOI:10.1021/ma801962v
Highly efficient, rapid, and reversible chain transfer between active transition-metal-based propagating centers derived from {Cp*Hf(Me)[N(Et)C(Me)N(Et)]}[B(C6F5)4] (Cp* = η5-C5Me5) (1a) or {Cp*Hf(Me)[N(Et)C(Me)N(Et)]}[B(C6F5)3Me] (1b) and multiple equivalents of dialkylzinc (ZnR2) acting as “surrogate” chain-growth sites has been achieved for establishing the living coordinative chain-transfer polymerization (CCTP) of ethene, α-olefins, and α,ω-nonconjugated dienes and living CCTP copolymerization of ethene with α-olefins and α,ω-nonconjugated dienes. These living CCTP processes not only provide a work-around solution to the “one chain per metal” cap on product yield currently limiting traditional living coordination polymerization of ethene and α-olefins but, in addition, provide access to practical volumes of a variety of unique new classes of precision polyolefins of tunable molecular weights and very narrow polydispersity (Mw/Mn ≤ 1.1).
Co-reporter:Chaiwat Engtrakul and Lawrence R. Sita
Organometallics 2008 Volume 27(Issue 5) pp:927-937
Publication Date(Web):February 8, 2008
DOI:10.1021/om700807x
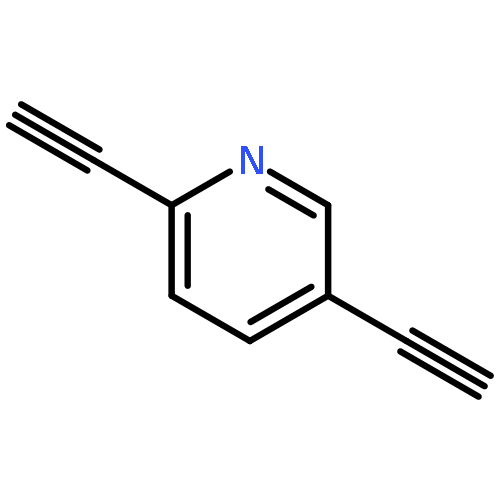
Regiospecific synthetic methods have been developed for the assembly of unsymmetric conjugated molecular frameworks containing 2,5-diethynylpyridyl- and 2,5-diethynylpyridinium-linked diferrocene structures and possessing either mono- or dithioacetate end-groups that are suitable for chemisorption onto Au(111) substrates after conversion to the corresponding thiol derivatives. Electronic spectra and solution electrochemistry of these and model compounds establish the electron-withdrawing character of a 2,5-dimethoxyphenylethynyl substituent on ferrocene that serves to shift the Fe(II)/Fe(III) redox couple to higher potentials. Further, while the unsymmetric nature of the 2,5-diethynylpyridyl bridge in 3 does not differentially perturb the redox couples of the two ferrocenes (ΔE1/2 < 10 mV), upon methylation, the corresponding pyridinium moiety of 4 now produces a large separation in the two redox potentials (ΔE1/2 = 190 mV). For the two regioisomeric monothioacetate compounds bearing a terminal 2,5-diethynylpyridyl-linked diferrocene unit, 5 and 6 (and their respective pyridinium counterparts, 7 and 8), redox potentials of the two ferrocenes are found to be either widely separated or similar in value depending upon the added influence of the 2,5-dimethoxyphenylethynyl group (e.g., ΔE1/2 = 310 mV in 7 vs ∼50 mV in 8).
Co-reporter:Matthew B. Harney;Yonghui Zhang Dr.
Angewandte Chemie 2006 Volume 118(Issue 15) pp:
Publication Date(Web):9 MAR 2006
DOI:10.1002/ange.200600027
Taktik in der Polymerisation: Das Molekulargewicht sowie die Zahl, Länge und Position eines jeden Stereoblocktyps in einem Polypropen wurden so programmiert, dass diskrete, isotaktisch/ataktische Stereoblockmikrostrukturen resultierten. Mehrere dieser gezielt entworfenen Stereoblockarchitekturen führen zu thermoplastischen Elastomeren (siehe Bild; weiße und rote Blöcke stehen für ataktische bzw. isotaktische Blöcke).
Co-reporter:Matthew B. Harney, Yonghui Zhang,Lawrence R. Sita
Angewandte Chemie International Edition 2006 45(15) pp:2400-2404
Publication Date(Web):
DOI:10.1002/anie.200600027
Co-reporter:Matthew B. Harney;Yonghui Zhang
Angewandte Chemie 2006 Volume 118(Issue 37) pp:
Publication Date(Web):21 AUG 2006
DOI:10.1002/ange.200601616
Ein neuer Blick auf Polypropylen: Indem der lebende Charakter der Propylenpolymerisation in einem genau definierten Zwei-Zustände-System genutzt wurde, gelang die Synthese einer Vielzahl an neuen stereogradienten Polypropylenmaterialien mit mikrostruktureller stereochemischer Homogenität aus einem einzigen Zirconocen-basierten Katalysator.
Co-reporter:Matthew B. Harney;Yonghui Zhang
Angewandte Chemie International Edition 2006 Volume 45(Issue 37) pp:
Publication Date(Web):21 AUG 2006
DOI:10.1002/anie.200601616
A new slant on polypropene: By taking advantage of the living nature of propene polymerization in a well-defined two-state system, an unlimited variety of polypropene microstructures, including a new stereogradient structure, can be prepared with stereochemical microstructural homogeneity.
Co-reporter:Denis A. Kissounko;Yonghui Zhang;Matthew B. Harney;Lawrence R. Sita
Advanced Synthesis & Catalysis 2005 Volume 347(Issue 2-3) pp:
Publication Date(Web):14 FEB 2005
DOI:10.1002/adsc.200404266
The dialkyl(monocyclopentadienyl)hafnium acetamidinates, Cp*Hf(R)2[N(Et)C(Me)N(t-Bu)] [Cp*=η5-C5Me5, R=Me (3a) and i-Bu (3b)], were prepared through either ‘one–pot’ carbodiimide insertion or two-step salt elimination protocols starting with commercially available Cp*HfCl3. Protonolysis of 3a and 3b with [PhNHMe2][B(C6F5)4] in chlorobenzene produced the cationic complexes, {[Cp*Hf(R)[N(Et)C(Me)N(t-Bu)]}[B(C6F5)4] [R=Me (4a) and i-Bu (4b)], which were characterized in solution. Compounds 4a and 4b were evaluated as initiators for the stereospecific living Ziegler–Natta polymerization of 1-hexene. These results reveal that, while an extremely high level of stereoselectivity can be achieved to produce isotactic poly(1-hexene) in a living fashion, the rate constant for polymerization, kp, using either 4a or 4b, is ~60 times less than that of the analogous zirconium initiators. Finally, upon substoichiometric activation of 3a with [PhNHMe2][B(C6F5)4] in a 2 : 1 ratio, degenerative transfer living Ziegler–Natta polymerization of 1-hexene can be accomplished to produce atactic poly(1-hexene).

![Silane,[[2,5-dimethoxy-4-[(trimethylsilyl)ethynyl]phenyl]ethynyl]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/873500/873464-11-0.png)
![Silane,[[2,5-dimethoxy-4-[(trimethylsilyl)ethynyl]phenyl]ethynyl]tris(1-methylethyl)-](http://img.cochemist.com/ccimg/873500/873464-11-0_b.png)


























![Benzenemethanamine, N-[(1,1-dimethylethyl)carbonimidoyl]-](http://img.cochemist.com/ccimg/1300/1205-86-3.png)
![Benzenemethanamine, N-[(1,1-dimethylethyl)carbonimidoyl]-](http://img.cochemist.com/ccimg/1300/1205-86-3_b.png)
![Cyclohexanamine, N-[(1,1-dimethylethyl)carbonimidoyl]-](http://img.cochemist.com/ccimg/1300/1202-53-5.png)
![Cyclohexanamine, N-[(1,1-dimethylethyl)carbonimidoyl]-](http://img.cochemist.com/ccimg/1300/1202-53-5_b.png)