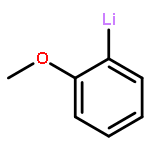
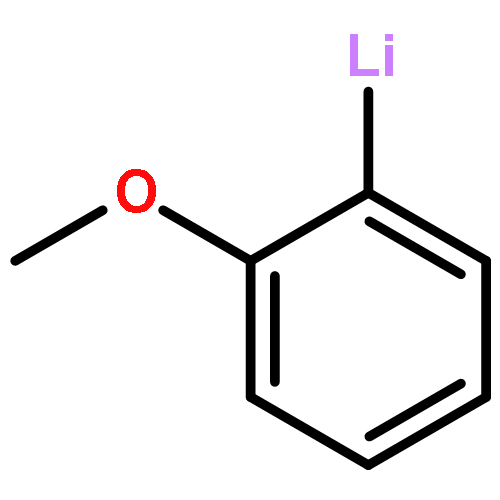
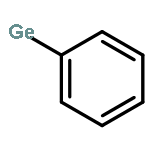
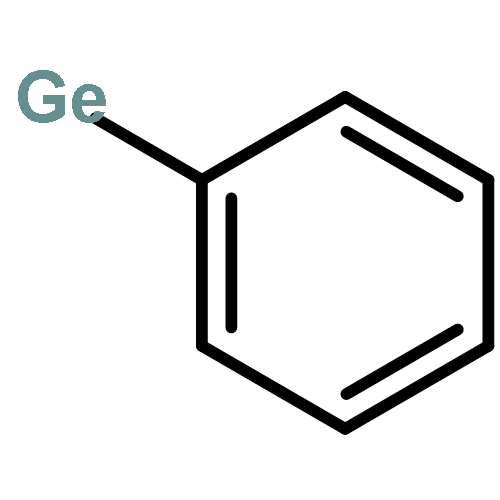
Co-reporter:Brittany J. Barrett and Vlad M. Iluc
Inorganic Chemistry July 21, 2014 Volume 53(Issue 14) pp:
Publication Date(Web):June 24, 2014
DOI:10.1021/ic500549z
The coordination chemistry of a neutral tPCH═CHP pincer (tPCH═CHP = 2,2′-bis(di-iso-propylphosphino)-trans-stilbene) with metals that form stable complexes in the +1 oxidation state was studied and (tPCH═CHP)CoCl, (tPCH═CHP)CoCl(CO), (tPCH═CHP)RhCl, (tPCH═CHP)Cu(OTf), [(tPCH═CHP)Cu][PF6], and [(tPCH═CHP)Ag][PF6] were synthesized and characterized. In order to determine whether the coordination mode is dependent on the oxidation state of the metal, some +2 metal complexes, (tPCH═CHP)CoCl2 and (tPCH═CHP)FeBr2, were also investigated. The coordination of the olefinic backbone is not observed in (tPCH═CHP)FeBr2, (tPCH═CHP)CoCl2, (tPCH═CHP)Cu(OTf), or [(tPCH═CHP)Ag][PF6], but η2-coordination is present in [(tPCH═CHP)CoCl][BArF4], [(tPCH═CHP)FeBr][BArF4], (tPCH═CHP)CoCl, (tPCH═CHP)CoCl(CO), (tPCH═CHP)RhCl, and [(tPCH═CHP)Cu][PF6]. Cobalt(II), iron(II), and copper(I) formed complexes with the ligand in both coordination modes. All metal complexes were characterized by multinuclei NMR spectroscopy, X-ray crystallography, and elemental analysis.
Co-reporter:Ama M. Holland;Allen G. Oliver;Vlad M. Iluc
Acta Crystallographica Section C 2017 Volume 73(Issue 7) pp:569-574
Publication Date(Web):2017/07/01
DOI:10.1107/S2053229617009287
The structure of a pincer ligand consists of a backbone and two `arms' which typically contain a P or N atom. They are tridentate ligands that coordinate to a metal center in a meridional configuration. A series of three iron complexes containing the pyrrole-based PNP pincer ligand 2,5-bis[(diisopropylphosphanyl)methyl]pyrrolide (PNpyrP) has been synthesized. These complexes are possible precursors to new iron catalysts. {2,5-Bis[(diisopropylphosphanyl)methyl]pyrrolido-κ3P,N,P′}carbonylchlorido(trimethylphosphane-κP)iron(II), [Fe(C18H34NP2)Cl(C3H9P)(CO)] or [Fe(PNpyrP)Cl(PMe3)(CO)], (I), has a slightly distorted octahedral geometry, with the Cl and CO ligands occupying the apical positions. {2,5-Bis[(diisopropylphosphanyl)methyl]pyrrolido-κ3P,N,P′}chlorido(pyridine-κN)iron(II), [Fe(C18H34NP2)Cl(C5H5N)] or [Fe(PNpyrP)Cl(py)] (py is pyridine), (II), is a five-coordinate square-pyramidal complex, with the pyridine ligand in the apical position. {2,5-Bis[(diisopropylphosphanyl)methyl]pyrrolido-κ3P,N,P′}dicarbonylchloridoiron(II), [Fe(C18H34NP2)Cl(CO)2] or [Fe(PNpyrP)Cl(CO)2], (III), is structurally similar to (I), but with the PMe3 ligand replaced by a second carbonyl ligand from the reaction of (II) with CO. The two carbonyl ligands are in a cis configuration, and there is positional disorder of the chloride and trans carbonyl ligands.
Co-reporter:Julie A. Kessler;Vlad M. Iluc
Dalton Transactions 2017 vol. 46(Issue 36) pp:12125-12131
Publication Date(Web):2017/09/19
DOI:10.1039/C7DT02784B
The reaction between [(PNpyrP)NiCl] (1, PNpyrP = 2,5-bis((di-iso-propylphosphino)-methyl)-1H-pyrrolide) and TlPF6 in the presence of a monodentate phosphine ligand led to cationic nickel phosphine and phosphite complexes, [(PNpyrP)Ni(PHPh2)][PF6] (2), [(PNpyrP)Ni(PMe3)][PF6] (3), and [(PNpyrP)Ni{P(OMe)3}][PF6] (4). Compound 2 can be deprotonated resulting in the generation of a terminal phosphido complex, [(PNpyrP)Ni(PPh2)] (5). When 3 is subjected to a base, a methyl proton of PMe3 is abstracted to afford [(PNpyrP)Ni(CH2PMe2)] (6), containing a methylene bridge between Ni and the external phosphine. Compounds 2–6 were characterized by single crystal X-ray diffraction in addition to multi-nuclear NMR spectroscopy and elemental analysis.
Co-reporter:Brittany J. Barrett and Vlad M. Iluc
Organometallics 2017 Volume 36(Issue 3) pp:
Publication Date(Web):January 20, 2017
DOI:10.1021/acs.organomet.6b00924
Group 10 metal carbenes are proposed in catalytic transformations; however, their isolation remains difficult without the presence of a heteroatom donor. The adaptable cis and trans coordinating ligand PterP (1,2-bis(2-(diisopropylphosphino)phenyl)benzene) is key in stabilizing two-coordinate palladium and platinum(0) precursors. Reacting these precursors with di-p-tolyldiazomethane ((p-tol)2CN2) leads to the formation of the unprecedented trigonal-planar diarylcarbenes [(PterP)M═C(p-tol)2] (M = Pd, Pt), upon transformation of the trans coordinating ligand into a wide-bite, cis-coordinating ligand. Both palladium and platinum diarylcarbenes were characterized by multinuclear NMR spectroscopy. The unusual stability of the platinum analogue allowed its characterization via X-ray crystallography. Furthermore, the reactivity of the palladium and platinum diarylcarbenes with Ph2SiH2 and CH3I was investigated.
Co-reporter:Brittany J. Barrett, Vlad M. Iluc
Inorganica Chimica Acta 2017 Volume 460(Volume 460) pp:
Publication Date(Web):24 April 2017
DOI:10.1016/j.ica.2016.08.035
•Interactions between a neutral tridentate ligand and palladium are investigated.•Hemilability is observed upon oxidative addition to Pd(0).•Hydrogen atom reservoir behavior is observed for the ligand backbone.The tridentate palladium(0) compound [(cPCMeCMeP)Pd] ((Z)-2,3-bis(2-di-iso-propylphosphinephenyl)-2-butene) was synthesized and its oxidative addition products with CH3I, C6H5I, Ph2SiH2, and HCl were characterized. Hemilability of the olefinic backbone is observed in all cases and, excluding Ph2SiH2, results in trans palladium(II) species. Oxidative addition of Ph2SiH2 results in a fluxional species due to the reversibility of the SiH bond activation. Reactivity with HCl led to the oxidative addition of the acid, resulting in the formation of a clorohydride complex, [(cPCMeCMeP)PdHCl], followed by a irreversible insertion of the olefin into the PdH bond resulting in [(PCMeCHMeP)PdCl]. Additionally, dehydrohalogenation was achieved with [(cPCMeCMeP)PdCl2] resulting in [(PCMeC(CH2)P)PdCl]. All metal complexes were characterized by multi-nuclei NMR spectroscopy, X-ray crystallography, and elemental analysis.The hemilability and the reactivity of the olefinic backbone of a chelate ligand was studied in several palladium complexes.Download high-res image (88KB)Download full-size image
Co-reporter:Peng Cui, Melissa R. Hoffbauer, Mariya Vyushkova and Vlad M. Iluc
Chemical Science 2016 vol. 7(Issue 7) pp:4444-4452
Publication Date(Web):24 Mar 2016
DOI:10.1039/C6SC00948D
Heterobimetallic Pd–K carbenes featuring Pd–Ccarbene–K moieties were synthesized via an unprecedented sequential substitution/reduction reaction from a radical precursor, [{PC˙(sp2)P}tBuPdI] ([PC(sp2)P]tBu = bis[2-(di-iso-propylphosphino)-4-tert-butylphenyl]methylene). Polymeric structures were observed in the solid state for the heterobimetallic compounds that can be interrupted in the presence of a donor solvent.
Co-reporter:Cezar C. Comanescu and Vlad M. Iluc
Chemical Communications 2016 vol. 52(Issue 58) pp:9048-9051
Publication Date(Web):21 Jan 2016
DOI:10.1039/C5CC09468B
The reactivity of two nucleophilic palladium carbenes, [PC(sp2)P]Pd(PMe3) and [PC(sp2)P]Pd(PPh3), where [PC(sp2)P] = bis[2-(di-iso-propylphosphino)phenyl]methylene, toward the E–H bond activation of Ph4−nEHn (E = Si, Ge; n = 1–3) and pinacolborane (HBpin) is discussed. Unlike previous reports, both types of isomer species, hydride [PC(EHn−1Ph4−n)P]PdH or [PC(Bpin)P]PdH and silyl/germyl [PC(H)P]Pd(EHn−1Ph4−n), were observed depending on the substrate and the phosphine ligand, showing that the polarity of the Pd–C bond can be tuned by the phosphine substituents.
Co-reporter:Peng Cui, Dominic C. Babbini and Vlad M. Iluc
Dalton Transactions 2016 vol. 45(Issue 24) pp:10007-10016
Publication Date(Web):17 Mar 2016
DOI:10.1039/C6DT00303F
Iridium PCsp3P complexes featuring a novel bis(2-diphenylphosphinophenyl)-2-pyridylmethane ligand (PCPyHP) are reported. C–H activation reactions between the dihydride complex [(PCPyP)Ir(H)2] and tetrahydrofuran or methyl tert-butyl ether in the presence of a hydrogen acceptor, norbornene (NBE), at ambient temperature led exclusively to the hydrido oxyalkyl complexes, [(PCPyP)IrH(C4H7O)] and [(PCPyP)IrH(CH2OtBu)], respectively. The internal pyridine donor is important and stabilizes these species by coordination to the iridium center. The coordination of pyridine to the iridium center is labile, however, and its dissociation occurs in the presence of a suitable substrate, as demonstrated by the intramolecular nucleophilic attack of pyridine on a vinylidene intermediate generated from PhCCH.
Co-reporter:Sean P. Vilanova and Vlad M. Iluc
Organometallics 2016 Volume 35(Issue 12) pp:2110-2123
Publication Date(Web):June 14, 2016
DOI:10.1021/acs.organomet.6b00333
The synthesis of the N-aryl-substituted PNP pro-ligands H(PNnaphP) (N-di(2-diisopropylphosphine-4-methylphenyl)naphthylamine) and H(PNtolP) (N-di(2-diisopropylphosphine-4-methylphenyl)-o-tolylamine) is reported. The corresponding iridium(III) complexes, [(PNnaphP)Ir(H)Cl], [(o-methyl-PNtolP)Ir(H)Cl], [(o-aryl-PNtolP)Ir(H)Cl], [(PNnaphP)Ir(H)2], [(o-methyl-PNtolP)Ir(H)2], and [(o-aryl-PNtolP)Ir(H)2], were also synthesized and structurally characterized, along with reaction intermediates, demonstrating various ligand coordination modes.
Co-reporter:Peng Cui and Vlad M. Iluc
Chemical Science 2015 vol. 6(Issue 12) pp:7343-7354
Publication Date(Web):25 Sep 2015
DOI:10.1039/C5SC02859K
Metal carbene complexes have been at the forefront of organic and organometallic synthesis and are instrumental in guiding future sustainable chemistry efforts. While classical Fischer and Schrock type carbenes have been intensely studied, compounds that do not fall within one of these categories have attracted attention only recently. In addition, applications of carbene complexes rarely take advantage of redox processes, which could open up a new dimension for their use in practical processes. Herein, we report an umpolung of a nucleophilic palladium carbene complex, [{PC(sp2)P}tBuPd(PMe3)] ({PC(sp2)P}tBu = bis[2-(di-iso-propylphosphino)-4-tert-butylphenyl]methylene), realized by successive one-electron oxidations that generated a cationic carbene complex, [{PC(sp2)P}tBuPdI]+, via a carbene radical, [{PC˙(sp2)P}tBuPdI]. An EPR spectroscopic study of [{PC˙(sp2)P}tBuPdI] indicated the presence of a ligand-centered radical, also supported by the results of reactions with 9,10-dihydroanthracene and PhSSPh. The cationic carbene complex shows electrophilic behavior toward nucleophiles such as NaH, pTolNHLi, PhONa, and PMe3, resulting from an inversion of the electronic character of the Pd–Ccarbene bond in [{PC(sp2)P}tBuPd(PMe3)]. The redox induced umpolung is reversible and unprecedented.
Co-reporter:C. C. Comanescu, M. Vyushkova and V. M. Iluc
Chemical Science 2015 vol. 6(Issue 8) pp:4570-4579
Publication Date(Web):18 May 2015
DOI:10.1039/C5SC01441G
A series of palladium(II) radical carbene complexes, [PC˙(sp2)P]PdI, [PC˙(sp2)P]PdBr, and [PC˙(sp2)P]PdCl (PC(sp3)H2P = bis[2-(di-iso-propylphosphino)-phenyl]methane), is described. Compound [PC˙(sp2)P]PdI dimerizes to {[PC(sp2)P]PdI}2 in the solid state, akin to the formation of Gomberg's dimer. While the bromo and the iodo derivatives could be obtained from the oxidation of [PC(sp2)P]Pd(PMe3) by the respective dihalogens, a halogen transfer reaction from CH2Cl2 was used for the formation of [PC˙(sp2)P]PdCl. The halogen transfer from CH2X2 (X = Cl, Br, I) could be used to obtain all three radical carbene palladium complexes and also allowed the isolation of [PC(CH2)P]Pd(PMe3), which is the result of methylene group transfer from CH2X2. Compound [PC(CH2)P]Pd(PMe3) was independently synthesized from [PC(CH3)HP]PdCl2, which contains a supporting ligand analogous to that of the radical carbene complexes but has one of the hydrogen atoms replaced by a methyl group. All three carbene radical species abstract a hydrogen from 9,10-dihydroanthracene or nBu3SnH.
Co-reporter:Peng Cui, Cezar C. Comanescu and Vlad M. Iluc
Chemical Communications 2015 vol. 51(Issue 28) pp:6206-6209
Publication Date(Web):03 Mar 2015
DOI:10.1039/C5CC00868A
The reactions of two nucleophilic palladium carbene complexes with the strong Lewis acid B(C6F5)3 afforded two zwitterionic products. One of them features a remote nucleophilic attack at the para-carbon of the supporting ligand, while the other indicates C–F activation of B(C6F5)3. Both behaviours are reminiscent of the reactivity of frustrated Lewis pairs due to the steric inaccessibility of the nucleophilic carbon center, but are unprecedented for transition metal carbene complexes. Furthermore, when those reactions are carried out in the presence of H2, products resulting from H2 splitting are observed.
Co-reporter:Dominic C. Babbini and Vlad M. Iluc
Organometallics 2015 Volume 34(Issue 13) pp:3141-3151
Publication Date(Web):June 24, 2015
DOI:10.1021/acs.organomet.5b00165
A series of iridium PCsp3P complexes based on bis(2-diisopropylphosphinophenyl)-2-anisoylmethane (PCanisHP) is reported. (PCanisP)Ir(H)Cl was generated from the C–H activation of the backbone by [Ir(COD)Cl]2 (COD = 1,5-cyclooctadiene), while the dihydride (PCanisP)Ir(H)2 was generated by hydride metathesis from (PCanisP)Ir(H)Cl. Both complexes are 18e octahedral complexes and water stable. The hemilability of the anisole tether was probed using CO and PMe3; multiple isomers, in which the anisole substituent was displaced, were generated, showing the flexibility of the ligand backbone. (PCanisP)Ir(H)2 showed deuterium incorporation in the hydride, backbone, and anisole positions upon moderate heating in C6D6. Both (PCanisP)Ir(H)Cl and (PCanisP)Ir(H)2 were precatalysts for transfer dehydrogenation of cyclooctane under moderate conditions.
Co-reporter:Cezar C. Comanescu and Vlad M. Iluc
Organometallics 2015 Volume 34(Issue 19) pp:4684-4692
Publication Date(Web):July 9, 2015
DOI:10.1021/acs.organomet.5b00414
The reactivity of a nucleophilic palladium carbene, [PC(sp2)P]Pd(PMe3) (1; [PC(sp2)P] = bis[2-(diisopropylphosphino)phenyl]methylene), toward the C–H bonds of CH3COCH3, CH3CN, Ph–CCH, fluorene, and 9,10-dihydroanthracene was investigated. All surveyed substrates reacted with 1. However, there was no detectable reaction of 1 with Ph2CH2. It is proposed that the pKa values of the studied C–H bonds govern their reactivity toward 1: our results show that substrates with a pKa higher than 29, such as Ph2CH2 (pKa = 32.2), do not react even with prolonged heating.
Co-reporter:Vlad M. Iluc ;Gregory L. Hillhouse
Journal of the American Chemical Society 2014 Volume 136(Issue 17) pp:6479-6488
Publication Date(Web):April 9, 2014
DOI:10.1021/ja501900j
The synthesis and characterization of two new carbene complexes, (dtbpe)Ni═CH(dmp) (1; dtbpe = 1,2-bis(di-tert-butylphosphino)ethane; dmp = 2,6-dimesitylphenyl) and (dippn)Ni═CH(dmp) (2; dippn = 1,8-bis(di-iso-propylphosphino)naphthalene), are described. Complexes 1 and 2 were isolated by photolysis of the corresponding side-bound diazoalkane complexes, exemplified by (dtbpe)Ni{η2-N2CH(dmp)} (3). The carbene complexes feature Ni–C distances that are short and Ni–C–C angles at the carbene carbon that are intermediate between 120° and 180° (155.7(3)° and 152.3(3)°, respectively). The difference between the two carbenes became obvious when their reactivity toward 1-electron oxidizing agents was studied: the oxidation of 1 led to an internal rearrangement and the formation of a nickel(I) alkyl [{κ2-P,C-di-tert-butylphosphino-di-tert-butyl-PCH(dmp)ethane}Ni][BArF4] (4), while the oxidation of 2 allowed the isolation of an unrearranged product, formulated as the cationic nickel(III) carbene complex[(dippn)Ni═CH(dmp)][BArF4] (6). Both oxidations are chemically reversible and the respective reductions lead to the neutral carbene complexes, 1 and 2.
Co-reporter:Julie A. Kessler and Vlad M. Iluc
Inorganic Chemistry 2014 Volume 53(Issue 23) pp:12360-12371
Publication Date(Web):November 11, 2014
DOI:10.1021/ic501600s
A PNP ligand, PNpyrP ((PNpyrP)H = 2,5-bis((di-iso-propylphosphino)methyl)pyrrole), which employs a pyrrole unit as a central anionic nitrogen donor, was designed. The corresponding group 10 metal chlorides as well as iridium and ruthenium compounds were isolated. In order to conduct this work, [(PNpyrP)Tl] and [(PNpyrP)Ag]2 were synthesized and characterized. The thallium and silver species were paramount in the formation of the iridium and ruthenium complexes, which could not be isolated using (PNpyrP)H or the corresponding lithium pyrrolide salt. Interestingly, the solid state molecular structure of [(PNpyrP)Tl] indicates that the metal center engages in an η2 intermolecular interaction with the backbone of a neighboring pyrrole molecule instead of the expected bonding to the phosphine arms.
Co-reporter:Cezar C. Comanescu and Vlad M. Iluc
Inorganic Chemistry 2014 Volume 53(Issue 16) pp:8517-8528
Publication Date(Web):August 7, 2014
DOI:10.1021/ic5010566
A series of diphosphine ligands iPr2P–C6H4–X–C6H4–PiPr2 (for ligand L1, X = CH2; for ligand L2, X = CH2CH2) was investigated to determine the preference for cis/trans coordination to palladium(0), palladium(II), and rhodium(I). Increasing the length of the bridging alkyl backbone from one to two carbons changes the geometry of the resulting palladium(II) complexes, with L1 coordinating preferentially cis, while L2 coordinates in a trans fashion. Coordination to Pd(0) leads to L1Pd(dba) and L2Pd(dba), in which both ligands accommodate a P–M–P angle close to 120°. L2 was found to coordinate cis in a rhodium(I) complex ([L2Rh(nbd)][BF4], where nbd = norbornadiene).
Co-reporter:Brittany J. Barrett and Vlad M. Iluc
Organometallics 2014 Volume 33(Issue 10) pp:2565-2574
Publication Date(Web):May 13, 2014
DOI:10.1021/om500256r
The coordination chemistry of 2,2′-bis(di-iso-propylphosphino)-trans-stilbene (tPCH═CHP) with group 10 metal centers in a variety of oxidation states is reported; different coordination modes were observed depending on the oxidation state of the metal. With metal centers in the 0 or +1 oxidation state ((tPCH═CHP)Ni, [(tPCH═CHP)Pd]2, (tPCH═CHP)NiCl, (tPCH═CHP)NiI), η2 coordination of the olefin occurs, whereas, with metals in the +2 oxidation state, C–H activation of the backbone, followed by rapid H–X reductive elimination, was observed, leading to an η1 coordination of the backbone in (tPC═CHP)MCl (M = Ni, Pd, Pt). Employing the methyl-substituted analogue, 2,2′-bis(di-iso-propylphosphino)-trans-diphenyl-1,2-dimethylethene (tPCMe═CMeP), forced an η2 coordination of the olefin in [(tPCMe═CMeP)NiCl]2[NiCl4]. The synthesis of the hydride complex (tPC═CHP)NiH was attempted, but, instead, led to the formation of (tPCH═CHP)Ni, indicating that the vinyl form of the backbone can function as a hydrogen acceptor. All metal complexes were characterized by multinuclei NMR spectroscopy, X-ray crystallography, and elemental analysis.
Co-reporter:Cezar C. Comanescu and Vlad M. Iluc
Organometallics 2014 Volume 33(Issue 21) pp:6059-6064
Publication Date(Web):October 7, 2014
DOI:10.1021/om500682s
Two formal palladium carbene complexes, [PC(sp2)P]Pd(PR3) (3: R = Me; 4: R = Ph) were isolated and characterized from [PC(sp3)H2P] ([PC(sp3)H2P] = bis[2-(di-isopropylphosphino)phenyl]methane, iPr2P-C6H4-CH2-C6H4-PiPr2). Structural studies and DFT calculations indicate that the interaction between palladium and carbon is best described as a single bond, associated with nucleophilic character at that carbon atom. The characteristics of 3 were probed by reactions with electrophiles (MeI), acids (MeOH and HCl), and para-toluidine.
Co-reporter:Peng Cui and Vlad M. Iluc
Chemical Science (2010-Present) 2015 - vol. 6(Issue 12) pp:NaN7354-7354
Publication Date(Web):2015/09/25
DOI:10.1039/C5SC02859K
Metal carbene complexes have been at the forefront of organic and organometallic synthesis and are instrumental in guiding future sustainable chemistry efforts. While classical Fischer and Schrock type carbenes have been intensely studied, compounds that do not fall within one of these categories have attracted attention only recently. In addition, applications of carbene complexes rarely take advantage of redox processes, which could open up a new dimension for their use in practical processes. Herein, we report an umpolung of a nucleophilic palladium carbene complex, [{PC(sp2)P}tBuPd(PMe3)] ({PC(sp2)P}tBu = bis[2-(di-iso-propylphosphino)-4-tert-butylphenyl]methylene), realized by successive one-electron oxidations that generated a cationic carbene complex, [{PC(sp2)P}tBuPdI]+, via a carbene radical, [{PC˙(sp2)P}tBuPdI]. An EPR spectroscopic study of [{PC˙(sp2)P}tBuPdI] indicated the presence of a ligand-centered radical, also supported by the results of reactions with 9,10-dihydroanthracene and PhSSPh. The cationic carbene complex shows electrophilic behavior toward nucleophiles such as NaH, pTolNHLi, PhONa, and PMe3, resulting from an inversion of the electronic character of the Pd–Ccarbene bond in [{PC(sp2)P}tBuPd(PMe3)]. The redox induced umpolung is reversible and unprecedented.
Co-reporter:C. C. Comanescu, M. Vyushkova and V. M. Iluc
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4579-4579
Publication Date(Web):2015/05/18
DOI:10.1039/C5SC01441G
A series of palladium(II) radical carbene complexes, [PC˙(sp2)P]PdI, [PC˙(sp2)P]PdBr, and [PC˙(sp2)P]PdCl (PC(sp3)H2P = bis[2-(di-iso-propylphosphino)-phenyl]methane), is described. Compound [PC˙(sp2)P]PdI dimerizes to {[PC(sp2)P]PdI}2 in the solid state, akin to the formation of Gomberg's dimer. While the bromo and the iodo derivatives could be obtained from the oxidation of [PC(sp2)P]Pd(PMe3) by the respective dihalogens, a halogen transfer reaction from CH2Cl2 was used for the formation of [PC˙(sp2)P]PdCl. The halogen transfer from CH2X2 (X = Cl, Br, I) could be used to obtain all three radical carbene palladium complexes and also allowed the isolation of [PC(CH2)P]Pd(PMe3), which is the result of methylene group transfer from CH2X2. Compound [PC(CH2)P]Pd(PMe3) was independently synthesized from [PC(CH3)HP]PdCl2, which contains a supporting ligand analogous to that of the radical carbene complexes but has one of the hydrogen atoms replaced by a methyl group. All three carbene radical species abstract a hydrogen from 9,10-dihydroanthracene or nBu3SnH.
Co-reporter:Peng Cui, Melissa R. Hoffbauer, Mariya Vyushkova and Vlad M. Iluc
Chemical Science (2010-Present) 2016 - vol. 7(Issue 7) pp:NaN4452-4452
Publication Date(Web):2016/03/24
DOI:10.1039/C6SC00948D
Heterobimetallic Pd–K carbenes featuring Pd–Ccarbene–K moieties were synthesized via an unprecedented sequential substitution/reduction reaction from a radical precursor, [{PC˙(sp2)P}tBuPdI] ([PC(sp2)P]tBu = bis[2-(di-iso-propylphosphino)-4-tert-butylphenyl]methylene). Polymeric structures were observed in the solid state for the heterobimetallic compounds that can be interrupted in the presence of a donor solvent.
Co-reporter:Peng Cui, Dominic C. Babbini and Vlad M. Iluc
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN10016-10016
Publication Date(Web):2016/03/17
DOI:10.1039/C6DT00303F
Iridium PCsp3P complexes featuring a novel bis(2-diphenylphosphinophenyl)-2-pyridylmethane ligand (PCPyHP) are reported. C–H activation reactions between the dihydride complex [(PCPyP)Ir(H)2] and tetrahydrofuran or methyl tert-butyl ether in the presence of a hydrogen acceptor, norbornene (NBE), at ambient temperature led exclusively to the hydrido oxyalkyl complexes, [(PCPyP)IrH(C4H7O)] and [(PCPyP)IrH(CH2OtBu)], respectively. The internal pyridine donor is important and stabilizes these species by coordination to the iridium center. The coordination of pyridine to the iridium center is labile, however, and its dissociation occurs in the presence of a suitable substrate, as demonstrated by the intramolecular nucleophilic attack of pyridine on a vinylidene intermediate generated from PhCCH.
Co-reporter:Cezar C. Comanescu and Vlad M. Iluc
Chemical Communications 2016 - vol. 52(Issue 58) pp:NaN9051-9051
Publication Date(Web):2016/01/21
DOI:10.1039/C5CC09468B
The reactivity of two nucleophilic palladium carbenes, [PC(sp2)P]Pd(PMe3) and [PC(sp2)P]Pd(PPh3), where [PC(sp2)P] = bis[2-(di-iso-propylphosphino)phenyl]methylene, toward the E–H bond activation of Ph4−nEHn (E = Si, Ge; n = 1–3) and pinacolborane (HBpin) is discussed. Unlike previous reports, both types of isomer species, hydride [PC(EHn−1Ph4−n)P]PdH or [PC(Bpin)P]PdH and silyl/germyl [PC(H)P]Pd(EHn−1Ph4−n), were observed depending on the substrate and the phosphine ligand, showing that the polarity of the Pd–C bond can be tuned by the phosphine substituents.
Co-reporter:Peng Cui, Cezar C. Comanescu and Vlad M. Iluc
Chemical Communications 2015 - vol. 51(Issue 28) pp:NaN6209-6209
Publication Date(Web):2015/03/03
DOI:10.1039/C5CC00868A
The reactions of two nucleophilic palladium carbene complexes with the strong Lewis acid B(C6F5)3 afforded two zwitterionic products. One of them features a remote nucleophilic attack at the para-carbon of the supporting ligand, while the other indicates C–F activation of B(C6F5)3. Both behaviours are reminiscent of the reactivity of frustrated Lewis pairs due to the steric inaccessibility of the nucleophilic carbon center, but are unprecedented for transition metal carbene complexes. Furthermore, when those reactions are carried out in the presence of H2, products resulting from H2 splitting are observed.