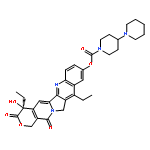
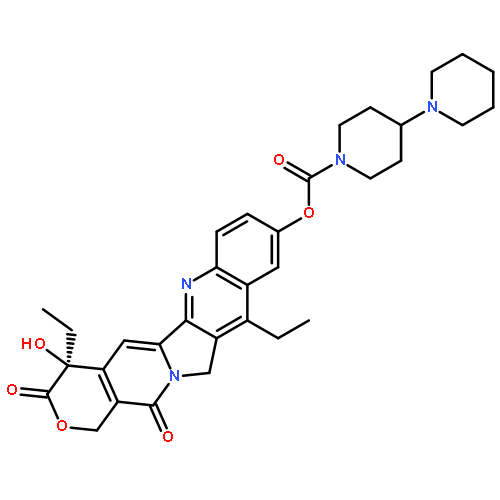
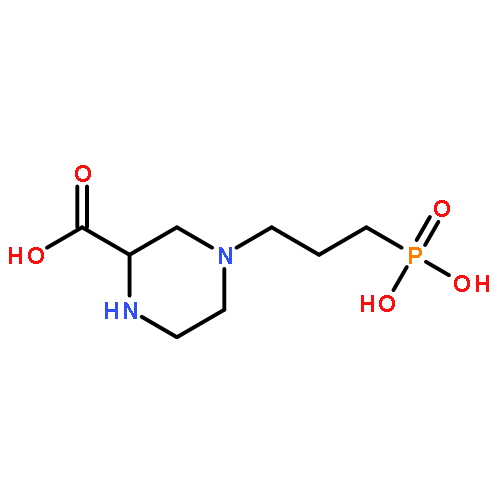
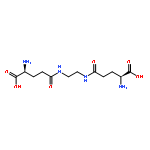
Co-reporter:Guixue Hou;Xiaomin Lou;Yulin Sun;Shaohang Xu;Jin Zi;Quanhui Wang;Baojin Zhou;Bo Han;Lin Wu;Xiaohang Zhao;Liang Lin
Journal of Proteome Research September 4, 2015 Volume 14(Issue 9) pp:3793-3803
Publication Date(Web):2017-2-22
DOI:10.1021/acs.jproteome.5b00438
We propose an efficient integration of SWATH with MRM for biomarker discovery and verification when the corresponding ion library is well established. We strictly controlled the false positive rate associated with SWATH MS signals and carefully selected the target peptides coupled with SWATH and MRM. We collected 10 samples of esophageal squamous cell carcinoma (ESCC) tissues paired with tumors and adjacent regions and quantified 1758 unique proteins with FDR 1% at protein level using SWATH, in which 467 proteins were abundance-dependent with ESCC. After carefully evaluating the SWATH MS signals of the up-regulated proteins, we selected 120 proteins for MRM verification. MRM analysis of the pooled and individual esophageal tissues resulted in 116 proteins that exhibited similar abundance response modes to ESCC that were acquired with SWATH. Because the ESCC-related proteins consisted of a high percentile of secreted proteins, we conducted the MRM assay on patient sera that were collected from pre- and postoperation. Of the 116 target proteins, 42 were identified in the ESCC sera, including 11 with lowered abundances postoperation. Coupling SWATH and MRM is thus feasible and efficient for the discovery and verification of cancer-related protein biomarkers.Keywords: biomarker; ESCC; MRM; SWATH;
Co-reporter:Yang Zhang;Guoquan Yan;Linhui Zhai;Shaohang Xu;Huali Shen;Jun Yao;Feifei Wu;Liqi Xie;Hailin Tang;Hongxiu Yu;Mingqi Liu;Pengyuan Yang;Ping Xu;Chengpu Zhang;Liwei Li;Cheng Chang;Ning Li;Songfeng Wu;Yunping Zhu;Quanhui Wang;Bo Wen;Liang Lin;Yinzhu Wang;Guiyan Zheng;Lanping Zhou;Haojie Lu;Fuchu He;Fan Zhong
Journal of Proteome Research January 4, 2013 Volume 12(Issue 1) pp:81-88
Publication Date(Web):2017-2-22
DOI:10.1021/pr300834r
Chromosome 8, a medium-length euchromatic unit in humans that has an extraordinarily high mutation rate, can be detected not only in evolution but also in multiple mutant diseases, such as tumorigenesis, and further invasion/metastasis. The Chromosome-Centric Human Proteome Project of China systematically profiles the proteomes of three digestive organs (i.e., stomach, colon, and liver) and their corresponding carcinoma tissues/cell lines according to a chromosome organizational roadmap. By rigorous standards, we have identified 271 (38.7%), 330 (47.1%), and 325 (46.4%) of 701 chromosome 8-coded proteins from stomach, colon, and liver samples, respectively, in Swiss-Prot and observed a total coverage rate of up to 58.9% by 413 identified proteins. Using large-scale label-free proteome quantitation, we also found some 8p deficiencies, such as the presence of 8p21–p23 in tumorigenesis of the above-described digestive organs, which is in good agreement with previous reports. To our best knowledge, this is the first study to have verified these 8p deficiencies at the proteome level, complementing genome and transcriptome data.Keywords: 8p deletion; chromosome 8; colon; liver; proteome; stomach; tumorigenesis;
Co-reporter:Xiuying Zhang;Haidan Sun;Sanjoy K. Paul;Quanhui Wang;Xiaomin Lou
Clinical Proteomics 2017 Volume 14( Issue 1) pp:19
Publication Date(Web):17 May 2017
DOI:10.1186/s12014-017-9154-0
The Xiaoke Pill containing Chinese herb extracts and Glibenclamide, is used in therapy for type 2 diabetes mellitus (T2DM), and is effective in reducing the risk of hypoglycemia and improving diabetes symptoms compared with Glibenclamide. We describe a quantitative proteomics project to measure the T2DM serum proteome response to the Xiaoke Pill and Glibenclamide.Based on a recently conducted 48-week clinical trial comparing the safety and efficacy of Glibenclamide (n = 400) and Xiaoke Pill (n = 400), after matching for age, sex, BMI, drug dose and whether hypoglycemia occurred, 32 patients were selected for the serum based proteomic analysis and divided into four groups (with/without hypoglycemia treated with Xiaoke Pill or Glibenclamide, n = 8 for each group). We screened the differential serum proteins related to treatments and the onset of hypoglycemia using the iTRAQ labeling quantitative proteomics technique. Baseline and follow-up samples were used.The quantitative proteomics experiments demonstrated that 25 and 21 proteins differed upon treatment with the Xiaoke Pill in patients without and with hypoglycemia, respectively, while 24 and 25 proteins differed upon treatment with Glibenclamide in patients without and with hypoglycemia, respectively. The overlap of different proteins between the patients with and without hypoglycemia given the same drug treatment was much greater than between the patients given different drug treatments.We conclude that the serum proteins response to the two different anti-diabetic drug treatments may serve as a sensitive biomarker for evaluation of the therapeutic effects and continue investigations into the mechanism.
Co-reporter:Yang Wang, Qiang Shan, Guixue Hou, Ju Zhang, Jian Bai, Xiaolei Lv, Yingying Xie, Huishan Zhu, Siyuan Su, Yang Li, Jin Zi, Liang Lin, Wenxiao Han, Xinhua Zhao, Hongying Wang, Ningzhi Xu, Lin Wu, Xiaomin Lou, Siqi Liu
Journal of Proteomics 2016 Volume 132() pp:31-40
Publication Date(Web):30 January 2016
DOI:10.1016/j.jprot.2015.11.013
•Dynamic TIF proteomics found 62 consecutively changed proteins related to CRC growth.•Targeted proteomics verified LRG1 and TUBB5 as potential CRC serum biomarkers.•Our study provides a promising pipeline to discover & verify tumor serum biomarkers.Quantitative proteomic analysis was performed using iTRAQ to discover colorectal cancer (CRC)-related proteins in tissue interstitial fluids (TIFs). A typical inflammation-related CRC mouse model was generated using azoxymethane–dextran sodium sulfate (AOM–DSS), and TIFs were collected from these mice in four stages during CRC development. Using stringent criteria, a total of 144 proteins displayed changes in their abundances during tumor growth, including 45 that consecutively increased, 17 that consecutively decreased and 82 that changed irregularly. Of these 144 proteins, 24 of the consecutively changed proteins were measured using MRM in individual TIF samples, and 18 were verified. Twelve proteins verified to be consecutively increased in TIFs were examined using MRM to evaluate changes in their abundance in individual mouse serum samples. The abundances of leucine-rich alpha-2-glycoprotein 1 (LRG1), tubulin beta-5 chain (TUBB5) and immunoglobulin J chain (IGJ) were significantly higher in CRC mice than in control mice. Using clinical samples and MRM, we further verified that LRG1 and TUBB5 are potential CRC serum biomarkers. These data demonstrate that coupling dynamic TIF proteomics with targeted serum proteomics in an animal model is a promising avenue for pursuing the discovery of tumor serum biomarkers.Biological significanceColorectal cancer (CRC) is one of the most dangerous diseases worldwide. However, few of CRC biomarkers possess satisfied specificity and sensitivity in clinical practices. Exploration of more CRC biomarkers, especially in serum, is an urgent and also a time-consuming campaign in the CRC study. Our study demonstrates that quantitatively evaluating the phase-dependent proteins in colonic tissue interstitial fluids from AOM–DSS mice is a feasible and effective way for exploration of the CRC-related proteins and the potential serum biomarkers. We identified two proteins, LRG1 and TUBB5, which may be practicable in human clinical samples as CRC serum biomarkers. To sum up, this study provides a novel angle to explore the critical factors in tumorigenesis and a new pipeline for potential serum biomarker discovery and verification.
Co-reporter:Jiao Guo; Yan Ren; Guixue Hou; Bo Wen; Feng Xian; Zhen Chen; Ping Cui; Yingying Xie; Jin Zi; Liang Lin; Song Wu; Zesong Li; Lin Wu; Xiaomin Lou
Journal of Proteome Research 2016 Volume 15(Issue 7) pp:2164-2177
Publication Date(Web):June 4, 2016
DOI:10.1021/acs.jproteome.6b00106
Urine is an ideal material to study the cancer-related protein biomarkers in bladder, whereas exploration to these candidates is confronting technique challenges. Herein, we propose a comprehensive strategy of searching the urine proteins related with bladder cancer. The strategy consists of three core combinations, screening the biomarker candidates in the secreted proteins derived from the bladder cancer cell lines and verifying them in patient urines, defining the differential proteins through two-dimensional electrophoresis (2DE) and isobaric tags for relative and absolute quantitation (iTRAQ) coupled with LC–MS/MS, and implementing quantitative proteomics of profiling and targeting analysis. With proteomic survey, a total of 700 proteins were found with their abundance of secreted proteins in cancer cell lines different from normal, while 87 proteins were identified in the urine samples. The multiple reaction monitoring (MRM)-based quantification was adapted in verifying the bladder cancer related proteins in individual urine samples, resulting in 10 differential urine proteins linked with the cancer. Of these candidates, receiver operating characteristic analysis revealed that the combination of CO3 and LDHB was more sensitive as the cancer indicator than other groups. The discovery of the bladder cancer indicators through our strategy has paved an avenue to further biomarker validation.
Co-reporter:Jiao Guo, Shaohang Xu, Xuanlin Huang, Lin Li, Congmin Zhang, Qingfei Pan, Zhen Ren, Ruo Zhou, Yan Ren, Jin Zi, Lin Wu, Jan Stenvang, Nils Brünner, Bo Wen, and Siqi Liu
Journal of Proteome Research 2016 Volume 15(Issue 11) pp:4047-4059
Publication Date(Web):July 26, 2016
DOI:10.1021/acs.jproteome.6b00387
A priority in solving the problem of drug resistance is to understand the molecular mechanism of how a drug induces the resistance response within cells. Because many cancer cells exhibit chromosome aneuploidy, we explored whether changes of aneuploidy status result in drug resistance. Two typical colorectal cancer cells, HCT116 and LoVo, were cultured with the chemotherapeutic drugs irinotecan (SN38) or oxaliplatin (QxPt), and the non- and drug-resistant cell lines were selected. Whole exome sequencing (WES) was employed to evaluate the aneuploidy status of these cells, and RNAseq and LC-MS/MS were implemented to examine gene expression at both mRNA and protein level. The data of gene expression was well-matched with the genomic conclusion that HCT116 was a near diploid cell, whereas LoVo was an aneuploid cell with the increased abundance of mRNA and protein for these genes located at chromosomes 5, 7, 12, and 15. By comparing the genomic, transcriptomic, and proteomic data, the LoVo cells with SN38 tolerance showed an increased genome copy in chromosome 14, and the expression levels of the genes on this chromosome were also significantly increased. Thus, we first observed that SN38 could impact the aneuploidy status in cancer cells, which was partially associated with the acquired drug resistance.Keywords: aneuploidy; chromosome; colorectal cancer; drug resistance; genome; proteome; transcriptome;
Co-reporter:Shaohang Xu; Ruo Zhou; Zhe Ren; Baojin Zhou; Zhilong Lin; Guixue Hou; Yamei Deng; Jin Zi; Liang Lin; Quanhui Wang; Xin Liu; Xun Xu; Bo Wen
Journal of Proteome Research 2015 Volume 14(Issue 12) pp:4976-4984
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.jproteome.5b00476
Considering the technical limitations of mass spectrometry in protein identification, the mRNAs bound to ribosomes (RNC-mRNA) are assumed to reflect the mRNAs participating in the translational process. The RNC-mRNA data are reasoned to be useful for appraising the missing proteins. A set of the multiomics data including free-mRNAs, RNC-mRNAs, and proteomes was acquired from three liver cancer cell lines. On the basis of the missing proteins in neXtProt (release 2014-09-19), the bioinformatics analysis was carried out in three phases: (1) finding how many neXtProt missing proteins have or do not have RNA-seq and/or MS/MS evidence, (2) analyzing specific physicochemical and biological properties of the missing proteins that lack both RNA-seq and MS/MS evidence, and (3) analyzing the combined properties of these missing proteins. Total of 1501 missing proteins were found by neither RNC-mRNA nor MS/MS in the three liver cancer cell lines. For these missing proteins, some are expected higher hydrophobicity, unsuitable detection, or sensory functions as properties at the protein level, while some are predicted to have nonexpressing chromatin structures on the corresponding gene level. With further integrated analysis, we could attribute 93% of them (1391/1501) to these causal factors, which result in the expression products scarcely detected by RNA-seq or MS/MS.
Co-reporter:Jin Zi, Shenyan Zhang, Ruo Zhou, Baojin Zhou, Shaohang Xu, Guixue Hou, Fengji Tan, Bo Wen, Quanhui Wang, Liang Lin, and Siqi Liu
Analytical Chemistry 2014 Volume 86(Issue 15) pp:7242
Publication Date(Web):June 26, 2014
DOI:10.1021/ac501828a
The strategy of sequential window acquisition of all theoretical fragment ion spectra (SWATH) is emerging in the field of label-free proteomics. A critical consideration for the processing of SWATH data is the quality of the ion library (or mass spectrometric reference map). As the availability of open spectral libraries that can be used to process SWATH data is limited, most users currently create their libraries in-house. Herein, we propose an approach to construct an expanded ion library using the data-dependent acquisition (DDA) data generated by fractionation proteomics. We identified three critical elements for achieving a satisfactory ion library during the iterative process of our ion library expansion, including a correction of the retention times (RTs) gained from fractionation proteomics, appropriate integrations of the fractionated proteomics into an ion library, and assessments of the impact of the expanded ion libraries to data mining in SWATH. Using a bacterial lysate as an evaluation material, we employed sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to fractionate the lysate proteins and constructed the expanded ion library using the fractionation proteomics data. Compared with the ion library built from the unfractionated proteomics, approximately 20% more peptides were extracted from the expanded ion library. The extracted peptides, moreover, were acceptable for further quantitative analysis.
Co-reporter:Yuan Wang, Hanfu Wei, Qin Pan, Zhaohui Wang, Rui Xing, Wenmei Li, Jun Zhang, Meng Ding, Jiao Guo, Lin Wu, Youyong Lu, Siqi Liu
Analytical Biochemistry 2012 Volume 430(Issue 1) pp:1-3
Publication Date(Web):1 November 2012
DOI:10.1016/j.ab.2012.07.019
High background interference during the antibody pair screening process is inevitable. In this study, we found that the high background was associated with heterophilic antibody interference introduced by the application of ascites-derived monoclonal antibodies when conducting large-scale antibody pair screening against different proteins. To eliminate antibody-associated heterophilic antibody interference, both blocking with mouse normal sera and antigen-mediated affinity chromatography were used, resulting in significant improvement in pairing performance and in antibody pair screening efficiency.
Co-reporter:Qiang Shan, Xiaomin Lou, Ting Xiao, Ju Zhang, Huiying Sun, Yanning Gao, Shujun Cheng, Lin Wu, Ningzhi Xu, Siqi Liu
Cancer Letters (1 January 2013) Volume 328(Issue 1) pp:160-167
Publication Date(Web):1 January 2013
DOI:10.1016/j.canlet.2012.08.019
Cancer/testis antigens (CTAs) are highly immunogenic in many tumors, especially in non-small cell lung cancer (NSCLC). A low-density protein microarray, which consisted of 72 CTAs and six non-CTAs, was used to screen for lung cancer-related autoantibodies. The CTA panel of NY-ESO-1, XAGE-1, ADAM29 and MAGEC1, had sensitivity and specificity values of 33% and 96%, respectively. When examined in a test set, this panel of markers had sensitivity and specificity values of 36% and 89%, respectively. This array of markers preferentially detected NSCLC, but did not detect breast cancer, and non-cancer lung disease.Highlights► It’s the first survey of autoantibodies in the serum of NSCLC using a comprehensive number of CTA. ► Our results greatly enrich our knowledge of the auto-immunity of CTAs in cancers and will definitely benefit future research in this area. ► By screening 377 sera from training and test set samples, we identified four CTAs are indicator of NSCLC.