Co-reporter:Kohei Iritani, Motoki Ikeda, Anna Yang, Kazukuni Tahara, Keiji Hirose, Jeffrey S. Moore, and Yoshito Tobe
Langmuir October 31, 2017 Volume 33(Issue 43) pp:12453-12453
Publication Date(Web):October 2, 2017
DOI:10.1021/acs.langmuir.7b03007
We present here hexagonal tiling using hexagonal phenylene-ethynylene and phenylene-butadiynylene macrocycles attached by alkyl ester groups, PEM-C6 and PBM-C8, respectively, or triethylene glycol ester groups, PEM-TEG and PBM-TEG, respectively, at each vertex of the macrocyclic periphery at the liquid/solid interface. In this study, we focused on the effects of macrocyclic core size and the chemical properties of side chains attached to macrocyclic cores as well as solute concentrations on the hexagonal geometry of self-assembled monolayers. STM observations at the 1,2,4-trichrolobenzene/graphite interface revealed that PEM-C6 formed a honeycomb structure by van der Waals interactions between the interdigitated alkyl chains. However, upon increasing solute concentration, it changed to more dense hexagonal structure (tentatively called loose hexagonal structure I). In contrast, PBM-C8 formed loose hexagonal structure II of a slightly different packing mode at low concentration, while at high concentration it formed a high-density hexagonal structure in which alkyl chains are not adsorbed on the surface (dense hexagonal structure). In the dense hexagonal structure, macrocyclic cores are linked by hydrogen bonds between the ester carbonyl oxygen and the aromatic hydrogen atoms of the neighboring macrocycles. The packing geometries of loose hexagonal structures of PEM-C6 and PBM-C8 are different due to the different distance between the attachment of the alkyl ester groups which are located in confined space. On the other hand, PEM-TEG and PBM-TEG formed dense hexagonal structures, similar to PBM-C8 at high concentration, with their TEG units not adsorbed on the surface.
Co-reporter:Yi Ren, Alexandr A. Banishev, Kenneth S. Suslick, Jeffrey S. Moore, and Dana D. Dlott
Journal of the American Chemical Society March 22, 2017 Volume 139(Issue 11) pp:3974-3974
Publication Date(Web):March 2, 2017
DOI:10.1021/jacs.7b00876
We describe ultrafast proton transfer in the ground electronic state triggered by the use of shock waves created by high-speed impacts. The emission of Nile Red (NR), a polarity sensing dye, was used to probe the effects of shock compression in a series of polymers, including polymer Brønsted bases blended with organic acid proton donors. NR undergoes a shock-induced red-shift due to an increase both in density and in polymer polarity. In blends with poly(4-vinylpyridine) (PVP) and phenol, NR showed an excess shock-induced red-shift with a distinct time dependence not present in controls that are incapable of proton transfer. The excess red-shift first appeared with 0.8 km·s–1 impacts. Occurring in ca. 10 ns, this NR red-shift was caused by the formation of an ion pair created by shock-triggered proton transfer from phenol to PVP.
Co-reporter:Timothy P. Moneypenny II, Nathan P. Walter, Zhikun Cai, Yu-Run Miao, Danielle L. Gray, Jordan J. Hinman, Semin Lee, Yang Zhang, and Jeffrey S. Moore
Journal of the American Chemical Society March 1, 2017 Volume 139(Issue 8) pp:3259-3259
Publication Date(Web):February 3, 2017
DOI:10.1021/jacs.7b00189
Porous materials provide a plethora of technologically important applications that encompass molecular separations, catalysis, and adsorption. The majority of research in this field involves network solids constructed from multitopic constituents that, when assembled either covalently or ionically, afford macromolecular arrangements with micro- or meso-porous apertures. Recently, porous solids fabricated from discrete organic cages have garnered much interest due to their ease of handling and solution processability. Although this class of materials is a promising alternative to network solids, fundamental studies are still required to elucidate critical structure–function relationships that govern microporosity. Here, we report a systematic investigation of the effects of building block shape-persistence on the porosity of molecular cages. Alkyne metathesis and edge-specific postsynthetic modifications afforded three organic cages with alkynyl, alkenyl, and alkyl edges, respectively. Nitrogen adsorption experiments conducted on rapidly crystallized and slowly crystallized solids illustrated a general trend in porosity: alkynyl > alkenyl > alkyl. To understand the molecular-scale origin of this trend, we investigated the short and long time scale molecular motions of the molecular cages using ab initio molecular dynamics (AIMD) and classical molecular dynamics (MD) simulations. Our combined experimental and computational results demonstrate that the microporosity of molecular cages directly correlates with shape persistence. These findings discern fundamental molecular requirements for rationally designing porous molecular solids.
Co-reporter:Ian D. Robertson, Leon M. Dean, Gabriel E. Rudebusch, Nancy R. Sottos, Scott R. White, and Jeffrey S. Moore
ACS Macro Letters June 20, 2017 Volume 6(Issue 6) pp:609-609
Publication Date(Web):May 24, 2017
DOI:10.1021/acsmacrolett.7b00270
Frontal ring-opening metathesis polymerization (FROMP) has potential for use in rapid fabrication of structural polymers. However, the high activity of the ruthenium catalyst used for FROMP has limited the working time to <1 h. We report the use of alkyl phosphites as inhibitors for Grubbs’ type catalysts to substantially extend working time. Subtle changes in alkyl phosphite structure are shown to impact both pot life and frontal velocity. Specifically, by varying phosphite structure and concentration, we are able to control pot life between 0.25 and 30 h while still allowing FROMP to proceed at velocities between 1 and 8 cm/min to yield fully cured thermoset polymers. These results are of interest for conventional ROMP synthesis and may open the way to new FROMP-based manufacturing possibilities.
Co-reporter:Elena C. Montoto, Gavvalapalli Nagarjuna, Jingshu Hui, Mark Burgess, Nina M. Sekerak, Kenneth Hernández-Burgos, Teng-Sing Wei, Marissa Kneer, Joshua Grolman, Kevin J. Cheng, Jennifer A. Lewis, Jeffrey S. Moore, and Joaquín Rodríguez-López
Journal of the American Chemical Society 2016 Volume 138(Issue 40) pp:13230-13237
Publication Date(Web):September 15, 2016
DOI:10.1021/jacs.6b06365
Versatile and readily available battery materials compatible with a range of electrode configurations and cell designs are desirable for renewable energy storage. Here we report a promising class of materials based on redox active colloids (RACs) that are inherently modular in their design and overcome challenges faced by small-molecule organic materials for battery applications, such as crossover and chemical/morphological stability. RACs are cross-linked polymer spheres, synthesized with uniform diameters between 80 and 800 nm, and exhibit reversible redox activity as single particles, as monolayer films, and in the form of flowable dispersions. Viologen-based RACs display reversible cycling, accessing up to 99% of their capacity and 99 ± 1% Coulombic efficiency over 50 cycles by bulk electrolysis owing to efficient, long-distance intraparticle charge transfer. Ferrocene-based RACs paired with viologen-based RACs cycled efficiently in a nonaqueous redox flow battery employing a simple size-selective separator, thus demonstrating a possible application that benefits from their colloidal dimensions. The unprecedented versatility in RAC synthetic and electrochemical design opens new avenues for energy storage.
Co-reporter:Maxwell J. Robb, Tae Ann Kim, Abigail J. Halmes, Scott R. White, Nancy R. Sottos, and Jeffrey S. Moore
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:12328-12331
Publication Date(Web):September 12, 2016
DOI:10.1021/jacs.6b07610
Transformation of naphthopyran into a colored merocyanine species in polymeric materials is achieved using mechanical force. We demonstrate that the mechanochemical reactivity of naphthopyran is critically dependent on the regiochemistry, with only one particular substitution pattern leading to successful mechanochemical activation. Two alternative regioisomers with different polymer attachment points are demonstrated to be mechanochemically inactive. This trend in reactivity is accurately predicted by DFT calculations, reinforcing predictive capabilities in mechanochemical systems. We rationalize the reactivity differences between naphthopyran regioisomers in terms of the alignment of the target C–O pyran bond with the direction of the applied mechanical force and its effect on mechanochemical transduction along the reaction coordinate.
Co-reporter:Semin Lee; Anna Yang; Timothy P. MoneypennyII
Journal of the American Chemical Society 2016 Volume 138(Issue 7) pp:2182-2185
Publication Date(Web):February 8, 2016
DOI:10.1021/jacs.6b00468
In dynamic covalent synthesis, kinetic traps are perceived as disadvantageous, hindering the system from reaching its thermodynamic equilibrium. Here we present the near-quantitative preparation of tetrahedral cages from simple tritopic precursors using alkyne metathesis. While the cages are the presumed thermodynamic sink, we experimentally demonstrate that the products no longer exchange their vertices once they have formed. The example reported here illustrates that kinetically trapped products may facilitate high yields of complex products from dynamic covalent synthesis.
Co-reporter:Kristin M. Hutchins, Nina M. Sekerak, and Jeffrey S. Moore
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:12336-12339
Publication Date(Web):September 5, 2016
DOI:10.1021/jacs.6b07742
Dispersions of particles onto which reactive groups are bound give rise to inhomogeneous concentrations that may afford fundamentally different chemical behavior compared to the same molecular species dissolved in homogeneous solution. An example is bimolecular reactivity of complementary-functionalized particles, whereby interparticle contact is expected to promote fast kinetics localized to the interface, while exhibiting essentially no reactivity elsewhere. Such materials may exhibit unique properties analogous to blood clotting and thereby be useful in self-healing applications. Here, we demonstrate a radical polymerization reaction whose initiation is controlled by the physical proximity of two complementary co-initiators bound to a substrate and/or polymer beads. Polymerization of the surrounding acrylate monomer only occurs when interfaces functionalized with dimethylaniline encounter interfaces bearing benzoyl peroxide. At the interface of the complementary-functionalized beads, polymerization affords a “clot-like” scaffold of beads and polymer. Interestingly, such a scaffold is only attained when the beads are in a quiescent state. These findings open the way to the design of spatially controlled dual initiator systems and novel self-healing strategies and motifs.
Co-reporter:Semin Lee, Etienne Chénard, Danielle L. Gray, and Jeffrey S. Moore
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:13814-13817
Publication Date(Web):October 10, 2016
DOI:10.1021/jacs.6b08752
Alkyne metathesis provided an efficient macrocyclization route to a cycloparaphenyleneacetylene derivative in high yield. The cavity size was suitably matched for C70 which was tightly bound in an induced-fit fashion. The strained alkynes enabled a copper-free, 3-fold azide–alkyne cycloaddition at 50 °C.
Co-reporter:Yi Ren, Semin Lee, James M. Christensen, Nikolay V. Plotnikov, Mark Burgess, Todd J. Martínez, Dana D. Dlott, and Jeffrey S. Moore
Chemistry of Materials 2016 Volume 28(Issue 18) pp:6446
Publication Date(Web):September 13, 2016
DOI:10.1021/acs.chemmater.6b02703
Co-reporter:Ke Yang, Zhikun Cai, Madhusudan Tyagi, Mikhail Feygenson, Joerg C. Neuefeind, Jeffrey S. Moore, and Yang Zhang
Chemistry of Materials 2016 Volume 28(Issue 9) pp:3227
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.chemmater.6b01429
As a compelling case of sensitive structure–property relationship, an odd–even effect refers to the alternating trend of physical or chemical properties on odd/even number of repeating structural units. In crystalline or semicrystalline materials, such odd–even effects emerge as manifestations of differences in the periodic packing patterns of molecules. Therefore, due to the lack of long-range order, such an odd–even phenomenon is not expected for dynamic properties in amorphous state. Herein, we report the discovery of a remarkable odd–even effect of dynamical properties in the liquid phase. In a class of glass-forming diammonium citrate ionic liquids, using incoherent quasi-elastic neutron scattering measurements, we measured the dynamical properties including diffusion coefficient and rotational relaxation time. These directly measured molecular dynamics showed pronounced alternating trends with increased number of methylene (−CH2−) groups in the backbone. Meanwhile, the structure factor S(Q) showed no long-range periodic packing of molecules, while the pair distribution function G(r) revealed subtle differences in the local molecular morphology. The observed dynamical odd–even phenomenon in liquids showed that profound dynamical changes originate from subtle local structural differences.
Co-reporter:Mark Burgess, Etienne Chénard, Kenneth Hernández-Burgos, Gavvalapalli Nagarjuna, Rajeev S. Assary, Jingshu Hui, Jeffrey S. Moore, and Joaquín Rodríguez-López
Chemistry of Materials 2016 Volume 28(Issue 20) pp:7362
Publication Date(Web):September 30, 2016
DOI:10.1021/acs.chemmater.6b02825
The design of chemically stable and electrochemically reversible redox active polymers (RAPs) is of great interest for energy storage technologies. Particularly, RAPs are new players for flow batteries relying on a size-exclusion based mechanism of electrolyte separation, but few studies have provided detailed molecular understanding of redox polymers in solution. Here, we use a systematic molecular design approach to investigate the impact of linker and redox-pendant electronic interactions on the performance of viologen RAPs. We used scanning electrochemical microscopy, cyclic voltammetry, bulk electrolysis, temperature-dependent absorbance, and spectroelectrochemistry to study the redox properties, charge transfer kinetics, and self-exchange of electrons through redox active dimers and their equivalent polymers. Stark contrast was observed between the electrochemical properties of viologen dimers and their corresponding polymers. Electron self-exchange kinetics in redox active dimers that only differ by their tether length and rigidity influences their charge transfer properties. Predictions from the Marcus–Hush theory were consistent with observations in redox active dimers, but they failed to fully capture the behavior of macromolecular systems. For example, polymer bound viologen pendants, if too close in proximity, do not retain chemical reversibility. In contrast to polymer films, small modifications to the backbone structure decisively impact the bulk electrolysis of polymer solutions. This first comprehensive study highlights the careful balance between electronic interactions and backbone rigidity required to design RAPs with superior electrochemical performance.
Co-reporter:Preston A. May, Nicholas F. Munaretto, Michael B. Hamoy, Maxwell J. Robb, and Jeffrey S. Moore
ACS Macro Letters 2016 Volume 5(Issue 2) pp:177
Publication Date(Web):January 17, 2016
DOI:10.1021/acsmacrolett.5b00855
A detailed understanding of the fundamental processes that govern mechanical transduction in covalent polymer mechanochemistry is essential to advance innovation in this field. In contrast to progress in the development of new mechanophores, the influence of polymer structure and composition on mechanochemical activity has received relatively little attention. In order to address this gap in knowledge, a continuous flow system with synchronous UV–vis absorption capabilities was designed to quantify the ultrasound-induced mechanical activation of a spiropyran mechanophore in real-time. Measurements of reaction kinetics with polymer tethers of varying repeating unit structure demonstrate that degree of polymerization is the key descriptor of mechanochemical activity, independent of molecular weight and pendant group constitution. These results have important implications for the rationalization of mechanochemical properties and the design of new mechanochemically active polymer systems.
Co-reporter:Yi Ren, Jaejun Lee, Kristin M. Hutchins, Nancy R. Sottos, and Jeffrey S. Moore
Crystal Growth & Design 2016 Volume 16(Issue 11) pp:6148
Publication Date(Web):September 29, 2016
DOI:10.1021/acs.cgd.6b01119
We report the single crystal structure and thermal properties of 1,2-bis(phenylethynyl)benzene (PEB), revealing that PEB forms a metastable liquid at rt, ca. 35 °C below its melting point. Accelerated nucleation of PEB from its supercooled state was induced with high reproducibility by a shock wave with ca. 15 ns duration and 1.2 GPa peak pressure. By conducting shock wave experiments with varying peak pressures, we observed a correlation between the frequency of accelerated nucleation and shock intensity. The generality of shock-induced nucleation for supercooled liquids was probed with other organic supercooled liquids bearing phenyl rings. However, accelerated nucleation after shock wave impact was only observed for PEB, possibly due to the low rotational energy barrier of the terminal phenyl rings.
Co-reporter:Maxwell J. Robb, Wenle Li, Ryan C. R. Gergely, Christopher C. Matthews, Scott R. White, Nancy R. Sottos, and Jeffrey S. Moore
ACS Central Science 2016 Volume 2(Issue 9) pp:598
Publication Date(Web):August 17, 2016
DOI:10.1021/acscentsci.6b00198
Microscopic damage inevitably leads to failure in polymers and composite materials, but it is difficult to detect without the aid of specialized equipment. The ability to enhance the detection of small-scale damage prior to catastrophic material failure is important for improving the safety and reliability of critical engineering components, while simultaneously reducing life cycle costs associated with regular maintenance and inspection. Here, we demonstrate a simple, robust, and sensitive fluorescence-based approach for autonomous detection of damage in polymeric materials and composites enabled by aggregation-induced emission (AIE). This simple, yet powerful system relies on a single active component, and the general mechanism delivers outstanding performance in a wide variety of materials with diverse chemical and mechanical properties.
Co-reporter:Etienne Chénard
The Journal of Physical Chemistry C 2016 Volume 120(Issue 16) pp:8461-8471
Publication Date(Web):March 31, 2016
DOI:10.1021/acs.jpcc.6b00858
Following the discovery of the redox-active 1,4-bis-BF3-quinoxaline complex, we undertook a structure–activity study with the objective to understand the active nature of the quinoxaline complex. Through systematic synthesis and characterization, we have compared complexes prepared from pyridine and pyrazine derivatives, as heterocyclic core analogues. This paper reports the structural requirements that give rise to the electrochemical features of the 1,4-bis-BF3-quinoxaline adduct. Using solution and solid-state NMR spectroscopy, the role of aromatic ring fusion and nitrogen incorporation in bonding and electronics was elucidated. We establish the boron atom location and its interaction with its environment from 1D and 2D solution NMR, X-ray diffraction analysis, and 11B solid-state NMR experiments. Crystallographic analysis of single crystals helped to correlate the boron geometry with 11B quadrupolar coupling constant (CQ) and asymmetry parameter (ηQ), extracted from 11B solid-state NMR spectra. Additionally, computations based on density functional theory were performed to predict electrochemical behavior of the BF3–heteroaromatic complexes. We then experimentally measured electrochemical potential using cyclic voltammetry and found that the redox potentials and CQ values are similarly affected by electronic changes in the complexes.
Co-reporter:Benjamin R. Bunes;Miao Xu;Yaqiong Zhang;Dustin E. Gross;Avishek Saha;Daniel L. Jacobs;Xiaomei Yang;Ling Zang
Advanced Materials 2015 Volume 27( Issue 1) pp:162-167
Publication Date(Web):
DOI:10.1002/adma.201404112
Co-reporter:Maxwell J. Robb
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:10946-10949
Publication Date(Web):August 21, 2015
DOI:10.1021/jacs.5b07345
Mechanical activation of a β-lactam mechanophore using ultrasound induces a formal [2 + 2] cycloelimination reaction producing ketene and imine functional groups—the reverse reaction of the Staudinger cycloaddition. This transformation is predicted by computational modeling and verified by kinetics and UV–vis absorption measurements as well as polymer end-group analysis using 1H and 13C NMR spectroscopy. Addition of the β-lactam motif to the current repertoire of covalent mechanophores coupled with the diverse reactivity of the ketene functional group provides a promising new platform for achieving materials capable of autonomic self-healing behavior in response to external forces.
Co-reporter:Ke Yang; Jaejun Lee; Nancy R. Sottos
Journal of the American Chemical Society 2015 Volume 137(Issue 51) pp:16000-16003
Publication Date(Web):December 10, 2015
DOI:10.1021/jacs.5b10721
Understanding shockwave-induced physical and chemical changes of impact-absorbing materials is an important step toward the rational design of materials that mitigate the damage. In this work, we report a series of network-forming ionic liquids (NILs) that possess an intriguing shockwave absorption property upon laser-induced shockwave. Microstructure analysis by X-ray scattering suggests nano-segregation of alkyl side chains and charged head groups in NILs. Further post-shock observations indicate changes in the low-Q region, implying that the soft alkyl domain in NILs plays an important role in absorbing shockwaves. Interestingly, we observe a shock-induced ordering in the NIL with the longest (hexyl) side chain, indicating that both nano-segregated structure and shock-induced ordering contribute to NIL’s shockwave absorption performance.
Co-reporter:Yaqiong Zhang, Miao Xu, Benjamin R. Bunes, Na Wu, Dustin E. Gross, Jeffrey S. Moore, and Ling Zang
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 14) pp:7471
Publication Date(Web):March 31, 2015
DOI:10.1021/acsami.5b01532
High-performance chemiresistive sensors were made using a porous thin film of single-walled carbon nanotubes (CNTs) coated with a carbazolylethynylene (Tg-Car) oligomer for trace vapor detection of nitroaromatic explosives. The sensors detect low concentrations of 4-nitrotoluene (NT), 2,4,6-trinitrotoluene (TNT), and 2,4-dinitrotoluene (DNT) vapors at ppb to ppt levels. The sensors also show high selectivity to NT from other common organic reagents at significantly higher vapor concentrations. Furthermore, by using Tg-Car/CNT sensors and uncoated CNT sensors in parallel, differential sensing of NT, TNT, and DNT vapors was achieved. This work provides a methodology to create selective CNT-based sensors and sensor arrays.Keywords: carbazole oligomer; carbon nanotube; chemiresistive vapor sensor; differential sensing; nitroaromatic explosive; thin film;
Co-reporter:Yi Ren and Jeffrey S. Moore
Polymer Chemistry 2015 vol. 6(Issue 48) pp:8325-8330
Publication Date(Web):12 Oct 2015
DOI:10.1039/C5PY01422K
Covalent conjugation of tetrathiafulvalene (TTF) moieties to macromolecular backbones combines unique properties of polymers, such as processability and high functional group density, with the outstanding redox properties of TTF to expand applications in organic electronics, chemical sensors, molecular switches, and nonlinear optical materials among others. Herein, we report the synthesis of a polymethacrylate backbone with 4-(hydroxymethyl)TTF (hmTTF) as the pendant group using post-polymerization modification of poly(N-hydroxysuccinimide methacrylate) to obtain the product polymer with a wide range of molecular weights (18 ≤ Mn ≤ 126 kDa) and up to ca. 70 mol% (76 wt%) TTF. The electrochemical and electronic properties of phmTTFMA are analogous to that of monomeric TTF. Film-forming phmTTFMA-based charge-transfer complexes (CTCs) with various electron acceptors including iodine, TCNQ, and chloranil showed an isotropic electrical conductivity. The magnitude of the conductivities of phmTTFMA-based CTCs is primarily affected by the electron affinity of the electron acceptors. The morphology of the amorphous phmTTFMA is strongly dependent on its electronic interactions with small-molecule electron acceptors. For example, a lamellar-like structure is observed when phmTTFMA is mixed with chloranil.
Co-reporter:Joshua M. Grolman, Bora Inci, and Jeffrey S. Moore
ACS Macro Letters 2015 Volume 4(Issue 4) pp:441
Publication Date(Web):April 1, 2015
DOI:10.1021/acsmacrolett.5b00194
We recently reported cationic cyclopolymerization of o-vinylbenzaldehydes initiated by boron trifluoride to generate acid-sensitive poly(o-(α-alkyl)vinylbenzaldehyde). Herein we report preparation of core–shell microcapsules (μCs) using flow-focusing microfluidic techniques with shells composed of poly(o-(α-methyl)vinylbenzaldehyde) (PMVB) that release their payload in response to dilute aqueous acid solution. Release profiles of encapsulated fluorescein isothiocyanate-labeled dextran from μCs are controlled by varying the proton concentration and shell-wall thickness. SEM studies indicate that the system’s unique reversible release mechanism involves porosity changes in the shell wall due to microcrack formation.
Co-reporter:Gavvalapalli Nagarjuna, Yi Ren, Jeffrey S. Moore
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3155-3159
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2015.01.011
Cyclization reaction pathways of aryl-substituted arenediynes depend on the interplay between the steric interactions and electronic properties of the aryl substituents. Herein, to further probe the impact of bulky and electron rich aryl substituents on the cyclization of arenediynes, anthracenyl-substituted arenediynes (1–3) with different ortho substituents were synthesized and their thermal, photo, and radical initiated cyclization reactions were studied.
Co-reporter:Hector Lopez Hernez;Seung-Kyun Kang;Olivia P. Lee;Suk-Won Hwang;Joshua A. Kaitz;Bora Inci;Chan Woo Park;Sangjin Chung;Nancy R. Sottos;John A. Rogers;Scott R. White
Advanced Materials 2014 Volume 26( Issue 45) pp:7637-7642
Publication Date(Web):
DOI:10.1002/adma.201403045
Co-reporter:Scott W. Sisco and Jeffrey S. Moore
Chemical Science 2014 vol. 5(Issue 1) pp:81-85
Publication Date(Web):02 Sep 2013
DOI:10.1039/C3SC52018H
Chiral arylene–ethynylene macrocycles (AEMs) were synthesized via alkyne metathesis-mediated depolymerization of BINOL-based polymers. Homochiral dimers are selectively obtained from metathesis of heterochiral polymers. Thermodynamic analysis and computational modeling suggests the homochiral self-sorting to be entropy-driven due to the greater symmetry of the homochiral dimers over the heterochiral dimer. This symmetry-controlled reaction is a novel approach to achieving high selectivity in dynamic covalent macrocycle synthesis. Importantly, the result describes a new paradigm in dynamic covalent chemistry that will enable efficient synthesis of new chiral architectures.
Co-reporter:Ian D. Robertson, Hector Lopez Hernandez, Scott R. White, and Jeffrey S. Moore
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 21) pp:18469
Publication Date(Web):October 16, 2014
DOI:10.1021/am5061596
Materials capable of rapidly modifying their physical properties in response to stimuli are desirable for on-demand deployment and adaptive engineering structures. Frontal polymerization is a self-propagating reaction that can quickly transform liquid reactants into solid products. In this contribution, we demonstrate that frontal polymerization enables facile, rapid stiffening of a vascular network embedded in a flexible matrix. Systematic variation of the vascular architecture shows that polymerization fronts in a polydimethylsiloxane (PDMS) matrix are self-propagating in channels as small as 838 μm and even when curves, branch points and converging geometries are present. After polymerization the composite structure was dramatically stiffened (up to 18 times the original Young’s modulus) based on tensile testing results. This work demonstrates the use of frontal polymerization as an efficient methodology for transforming flexible materials into functional supports or surfaces through endoskeletal stiffening.Keywords: frontal polymerization; microfluidic endoskeleton; rapid stiffening; shape fixing
Co-reporter:Charles E. Diesendruck, Lingyang Zhu and Jeffrey S. Moore
Chemical Communications 2014 vol. 50(Issue 87) pp:13235-13238
Publication Date(Web):30 Jul 2014
DOI:10.1039/C4CC03514C
We investigated the use of mechanical stress to bend carbon–carbon triple bonds. Formation of an isoquinoline after reaction with a benzyl azide trap points towards a nucleophilic addition mechanism, differentiating mechanochemical trans-bending of π bonds from the typical reactivity observed for cisoidal bending of triple bonds in strained cyclic alkynes.
Co-reporter:Joshua A. Kaitz, Catherine M. Possanza, Yang Song, Charles E. Diesendruck, A. Jolanda H. Spiering, E. W. Meijer and Jeffrey S. Moore
Polymer Chemistry 2014 vol. 5(Issue 12) pp:3788-3794
Publication Date(Web):18 Mar 2014
DOI:10.1039/C3PY01690K
Incorporation of supramolecular cross-linking motifs into low-ceiling temperature (Tc) polymers allows for the possibility of remendable polymeric networks and nanoparticles whose structure and chemical backbones can be dynamically modified or depolymerized as desired. Herein, we demonstrate the synthesis of phthalaldehyde–benzaldehyde copolymers bearing a pendant dimerizing 2-ureido-pyrimidinone (UPy) motif. The UPy moiety promotes single-chain polymeric nanoparticle formation through non-covalent cross-linking at intermediate concentrations and results in reversible polymer network formation at high concentrations. Furthermore, due to the low Tc polymer backbone within such macromolecules, the materials depolymerize to monomer under appropriate conditions. We envision that the synthesis of such depolymerizable, adaptive supramolecular polymeric materials may find use in materials capable of self-healing and remodeling as well as in triggered release applications or the development of nanoporous structures.
Co-reporter:Joshua A. Kaitz and Jeffrey S. Moore
Macromolecules 2014 Volume 47(Issue 16) pp:5509-5513
Publication Date(Web):August 6, 2014
DOI:10.1021/ma5013557
Aldehyde polymers have gained attention in recent decades as a class of stimuli-responsive materials capable of rapid, triggered depolymerization to monomer. Exploitation of the most prominent polyaldehydes for various solid-state applications, however, is limited by poor thermal and mechanical properties of the materials. To address these limitations, we pursued the copolymerization of ethyl glyoxylate, precursor to tacky polymers, with o-phthalaldehyde, precursor to brittle materials. Using NMR spectroscopy and MALDI-TOF mass spectrometry, we have discovered the surprising tendency of these sequences to alternate, resulting in alternating cyclic copolymers in certain feed ratios. We also report the ability to tailor the thermal properties of the solid copolymers by varying copolymer composition, enabling the selective tuning of copolymer glass transition and degradation temperatures to meet application demands. We envision that this copolymer system, which blends the properties of the tacky and brittle homopolymers, will find use as depolymerizable polyaldehydes for solid-state applications.
Co-reporter:Joshua A. Kaitz, Charles E. Diesendruck, and Jeffrey S. Moore
Macromolecules 2014 Volume 47(Issue 11) pp:3603-3607
Publication Date(Web):May 27, 2014
DOI:10.1021/ma500674c
We recently discovered that the cationic polymerization of o-phthalaldehyde generates cyclic poly(phthalaldehyde) in high yield, high molecular weight, and a high degree of cyclic purity. Given this surprising result, we pursued the cationic polymerization of ethyl glyoxylate to determine if the macrocyclization outcome is, in fact, a general trend of low ceiling temperature polyacetals. Using NMR spectroscopy, MALDI-TOF mass spectrometry, and triple detection GPC, we have uncovered divergent macrocyclization mechanisms in the cationic polymerization of ethyl glyoxylate. Backbiting is observed either via the backbone acetal or via the pendant ester to give disparate polymer products and unique polymer architectures. The favored route for cyclization is found to depend on both the monomer concentration and the initiating species. Understanding the underlying mechanisms of polymerization and the ability to rigorously control polymer structure has important implications for the design of new transient materials.
Co-reporter:Scott W. Sisco, Brandon M. Larson, and Jeffrey S. Moore
Macromolecules 2014 Volume 47(Issue 12) pp:3829-3836
Publication Date(Web):June 2, 2014
DOI:10.1021/ma500673x
The preparation of functional macrocycles via dynamic covalent chemistry (DCC) is an attractive synthetic strategy due to its thermodynamic control over the products. The use of alkyne metathesis has emerged as an efficient DCC method for synthesis of conjugated arylene-ethynylene macrocycles (AEMs), but the scope has been mostly limited to rigid and angle-persistent benzenoid-based structures. Introducing functional groups to macrocycle backbones increases flexibility by relaxing conformational constraints, which often leads to broad product distributions. Here we expand the scope of alkyne metathesis to functionalized macrocycles by systematically exploring how conformation and connectivity interplay to affect product distribution. With a divergent approach, we prepared a series of conjugated polymer analogues and synthesized the corresponding macrocycles via depolymerization. The importance of conformational constraints was reinforced by the results, but it was discovered that minimizing the monomer’s torsional axes provides high yields of functional macrocycles, even those with increased flexibility. We believe that this strategy is applicable in other areas of DCC and self-assembly to enable efficient preparation of organic materials with flexible functional groups.
Co-reporter:Preston A. May and Jeffrey S. Moore
Chemical Society Reviews 2013 vol. 42(Issue 18) pp:7497-7506
Publication Date(Web):11 Jan 2013
DOI:10.1039/C2CS35463B
Long chain polymers have a unique ability to become highly extended in elongational flow fields. The forces developed along the backbone give rise to scission of the chains near their center. Recently, this unique property of polymers has been adopted to explore new chemical transformations by embedding structural elements into the backbone designed to undergo site-specific bond cleavage, termed mechanophores. Experimental techniques to generate elongational flow fields exist in a variety of different arrangements and have been used to study polymer mechanochemistry in solution. This tutorial review will discuss progress in the field of polymer mechanochemistry as well as survey the techniques used to generate elongational flow fields. Ultrasonication will be highlighted as the technique that has been widely adopted to screen mechanophore reactivity in solution.
Co-reporter:Yuan Gao;Ana M. Saenz de Jubera;Benito J. Mariñas
Advanced Functional Materials 2013 Volume 23( Issue 5) pp:598-607
Publication Date(Web):
DOI:10.1002/adfm.201201004
Abstract
The modification of a commercial nanofiltration (NF) membrane (TFC-S) with shape-persistent dendritic molecules is reported. Amphiphilic aromatic polyamide dendrimers (G1–G3) are synthesized via a divergent approach and used for membrane active layer modification by direct percolation. The permeate samples collected from the percolation experiments are analyzed by UV-visible spectroscopy to monitor the influence of dendrimer generations on percolation behavior and active layer modification. Further characterization of modified membranes by Rutherford backscattering spectrometry and atomic force microscopy techniques reveals a relatively low-level accumulation of dendrimers inside the original TFC-S NF membrane active layer and subsequent formation of a coating of pure aramide dendrimers on top of the active layer. A PES-PVA ultrafiltration membrane is used as a control membrane support (without an NF active layer) showing that structural compatibility between the dendrimers and support plays an important role in the membrane modification process. The performance of the modified TFC-S membrane is evaluated on the basis of the rejection abilities for a variety of water contaminants having a range of molecular size and chemistry. As the water flux is inversely proportional to the thickness of the active layer, the amount of dendrimers deposited for specific contaminants are optimized to improve the solute rejection while maintaining high water flux.
Co-reporter:Helin Huang, Dustin E. Gross, Xiaomei Yang, Jeffrey S. Moore, and Ling Zang
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 16) pp:7704
Publication Date(Web):August 8, 2013
DOI:10.1021/am402420g
High dark electrical conductivity was obtained for a p-type organic nanofibril material simply through a one-step surface doping. The nanofibril composite thus fabricated has been proven robust under ambient conditions. The high conductivity, combined with the intrinsic large surface area of the nanofibers, enables development of chemiresistor sensors for trace vapor detection of amines, with detection limit down to sub-parts per billion range.Keywords: high conductivity; organic nanofiber; p-type materials; sensors; surface doping; vapor detection;
Co-reporter:Bora Inci, Pin-Nan Cheng, Kyle Beljanski, and Jeffrey S. Moore
ACS Macro Letters 2013 Volume 2(Issue 10) pp:935
Publication Date(Web):October 3, 2013
DOI:10.1021/mz4004908
The cationic cyclopolymerization of o-vinylbenzaldehydes initiated by boron trifluoride is described. Unlike the incomplete conversion of o-vinylbenzaldehyde (1) at 0 °C, α-methyl-substituted monomers (2) and (3) undergo cyclopolymerizations with complete conversions at −78 °C. On the other hand, α-phenyl-substituted monomer (4) generated indenyl alcohol (7) when subjected to cationic polymerization conditions. The proposed mechanism for o-(α-methyl)vinylbenzaldehyde polymerization explains the importance of reaction temperature for polymer formation. Resulting amorphous poly(o-(α-methyl)vinylbenzaldehyde (10) exhibited good thermal stability (Tonset = 340 °C) with a Tg of 153 °C. Polymer (10) is a brittle and glassy plastic with a storage modulus (E′) of 3 × 108 Pa and elongation at break of ∼3%.
Co-reporter:Joshua A. Kaitz and Jeffrey S. Moore
Macromolecules 2013 Volume 46(Issue 3) pp:608-612
Publication Date(Web):February 4, 2013
DOI:10.1021/ma302575s
End-capped poly(phthalaldehyde) [PPA] is a well-studied metastable polymer that has attracted interest due to its ease of synthesis and rapid depolymerization. PPA is limited, however, in the type of macromolecular architectures accessible, as functionalizable phthalaldehyde derivatives are not commercially available and their synthesis is cumbersome. To this end, a general route to phthalaldehyde–benzaldehyde copolymers was sought, as benzaldehyde comonomers with various pendant functionalities are readily available. It was found that copolymers are synthesized by an anionic initiated polymerization of phthalaldehyde and electron-deficient benzaldehydes. The comonomer reactivities are shown to be sensitive to the benzaldehyde electronics; the relative reactivity of phthalaldehyde–benzaldehyde comonomer pairs strongly correlate with the Hammett values of the benzaldehyde monomer. These copolymers are then further modified to yield cross-linked, degradable polymer networks in just a two-step sequence. Phthalaldehyde–benzaldehyde copolymers thus enable functionalization of metastable polymers that rapidly depolymerize upon exposure to acid, thereby facilitating the development of triggerable degradation of polymer networks.
Co-reporter:Joshua A. Kaitz, Charles E. Diesendruck, and Jeffrey S. Moore
Macromolecules 2013 Volume 46(Issue 20) pp:8121-8128
Publication Date(Web):October 4, 2013
DOI:10.1021/ma401744k
We recently reported the cationic polymerization of o-phthalaldehyde to macrocyclic poly(phthalaldehyde) polymers. Resubjecting the cyclic polymers to the polymerization conditions led to a redistribution of the polymer to a new cyclic structure consistent with thermodynamic equilibrium. We now report the synthesis of cyclic poly(phthalaldehyde) derivatives and demonstrate the scrambling of distinct homopolymer mixtures to copolymers under the cationic polymerization conditions. Homopolymer mixtures are found to rapidly redistribute, first to multiblock cyclic copolymers. With extended reaction time, random macrocyclic copolymers are obtained. Evolution of the microstructure was monitored by NMR spectroscopy, MALDI–TOF mass spectrometry, and gel permeation chromatography (GPC). The reported scrambling method leads to the rapid preparation of macrocyclic copolymers of high molecular weight with variable microstructure depending on reaction times and catalyst loadings.
Co-reporter:Scott W. Sisco
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9114-9117
Publication Date(Web):May 17, 2012
DOI:10.1021/ja303572k
Macrocyclic oligomers possessing direction-defining ester linkages were synthesized via metathesis of the nondirectional alkyne functional group. Alkyne metathesis is expected to scramble the relative orientation of adjacent ester groups, potentially leading to a complex mixture of macrocyclic products. We wondered whether a narrow product distribution would be achievable with a proper choice of the building block structure. Here we show that the shape of the building block determines whether the macrocyclic products are directionally uniform or scrambled. Specifically, two isomeric arylene-ethynylene polyesters afforded significantly different product distributions upon being subjected to depolymerization–macrocyclization. These results underscore the importance of learning how the shape and geometry of the building blocks affect the macrocyclization energy landscape.
Co-reporter:Charles E. Diesendruck ; Brian D. Steinberg ; Naoto Sugai ; Meredith N. Silberstein ; Nancy R. Sottos ; Scott R. White ; Paul V. Braun
Journal of the American Chemical Society 2012 Volume 134(Issue 30) pp:12446-12449
Publication Date(Web):July 9, 2012
DOI:10.1021/ja305645x
A novel mechanophore with acid-releasing capability is designed to produce a simple catalyst for chemical change in materials under mechanical stress. The mechanophore, based on a gem-dichlorocyclopropanated indene, is synthesized and used as a cross-linker in poly(methyl acrylate). Force-dependent rearrangement is demonstrated for cross-linked mechanophore samples loaded in compression, while the control shows no significant response. The availability of the released acid is confirmed by exposing a piece of insoluble compressed polymer to a pH indicator solution. The development of this new mechanophore is the first step toward force-induced remodeling of stressed polymeric materials utilizing acid-catalyzed cross-linking reactions.
Co-reporter:Yanke Che ; Dustin E. Gross ; Helin Huang ; Dongjiang Yang ; Xiaomei Yang ; Emre Discekici ; Zheng Xue ; Huijun Zhao ; Jeffrey S. Moore ;Ling Zang
Journal of the American Chemical Society 2012 Volume 134(Issue 10) pp:4978-4982
Publication Date(Web):February 16, 2012
DOI:10.1021/ja300306e
Development of simple, cost-effective, and sensitive fluorescence-based sensors for explosives implies broad applications in homeland security, military operations, and environmental and industrial safety control. However, the reported fluorescence sensory materials (e.g., polymers) usually respond to a class of analytes (e.g., nitroaromatics), rather than a single specific target. Hence, the selective detection of trace amounts of trinitrotoluene (TNT) still remains a big challenge for fluorescence-based sensors. Here we report the selective detection of TNT vapor using the nanoporous fibers fabricated by self-assembly of carbazole-based macrocyclic molecules. The nanoporosity allows for time-dependent diffusion of TNT molecules inside the material, resulting in further fluorescence quenching of the material after removal from the TNT vapor source. Under the same testing conditions, other common nitroaromatic explosives and oxidizing reagents did not demonstrate this postexposure fluorescence quenching; rather, a recovery of fluorescence was observed. The postexposure fluorescence quenching as well as the sensitivity is further enhanced by lowering the highest occupied molecular orbital (HOMO) level of the nanofiber building blocks. This in turn reduces the affinity for oxygen, thus allocating more interaction sites for TNT. Our results present a simple and novel way to achieve detection selectivity for TNT by creating nanoporosity and tuning molecular electronic structure, which when combined may be applied to other fluorescence sensor materials for selective detection of vapor analytes.
Co-reporter:Aniket Datar, Dustin E. Gross, Kaushik Balakrishnan, Xiaomei Yang, Jeffrey S. Moore and Ling Zang
Chemical Communications 2012 vol. 48(Issue 71) pp:8904-8906
Publication Date(Web):16 Jul 2012
DOI:10.1039/C2CC34127A
Large area uniform nanofibers have been fabricated from a hexameric arylene–ethynylene macrocycle (1) through in situ self-assembly on a glass substrate during solvent evaporation. The fibril morphology is controlled by the solvophilic core of 1, in conjunction with the interfacial interactions between the side chains of 1 and the substrate.
Co-reporter:Dustin E. Gross, Emre Discekici and Jeffrey S. Moore
Chemical Communications 2012 vol. 48(Issue 37) pp:4426-4428
Publication Date(Web):20 Mar 2012
DOI:10.1039/C2CC30701D
A dynamic combinatorial approach for the synthesis of arylene ethynylene macrocycles (AEMs) from linear polymers is described. By using readily available carbazolyl-ethynylene copolymers as starting materials we obtained a number of novel macrocycles that would be difficult to prepare by traditional methods.
Co-reporter:Hefei Dong, Aaron P. Esser-Kahn, Piyush R. Thakre, Jason F. Patrick, Nancy R. Sottos, Scott R. White, and Jeffrey S. Moore
ACS Applied Materials & Interfaces 2012 Volume 4(Issue 2) pp:503
Publication Date(Web):October 18, 2011
DOI:10.1021/am2010042
When heated, poly(lactic acid) (PLA) fibers depolymerize in a controlled manner, making them potentially useful as sacrificial fibers for microchannel fabrication. Catalysts that increase PLA depolymerization rates are explored and methods to incorporate them into commercially available PLA fibers by a solvent mixture impregnating technique are tested. In the present study, the most active catalysts are identified that are capable of lowering the depolymerization temperature of modified PLA fibers by ca. 100 °C as compared to unmodified ones. Lower depolymerization temperatures allow PLA fibers to be removed from a fully cured epoxy thermoset resin without causing significant thermal damage to the epoxy. For 500 μm diameter PLA fibers, the optimized treatment involves soaking the fibers for 24 h in a solvent mixture containing 60% trifluoroethanol (TFE) and 40% H2O dispersed with 10 wt % tin(II) oxalate and subsequent air-drying of the fibers. PLA fibers treated with this procedure are completely removed when heated to 180 °C in vacuo for 20 h. The time evolution of catalytic depolymerization of PLA fiber is investigated by gel permeation chromatography (GPC). Channels fabricated by vaporization of sacrificial components (VaSC) are subsequently characterized by scanning electron microscopy (SEM) and X-ray microtomography (Micro CT) to show the presence of residual catalysts.Keywords: composites; microvascular network; poly(lactic acid) (PLA); sacrificial fiber; thermal depolymerization;
Co-reporter:Ligui Li, Yanke Che, Dustin E. Gross, Helin Huang, Jeffrey S. Moore, and Ling Zang
ACS Macro Letters 2012 Volume 1(Issue 11) pp:1335
Publication Date(Web):October 31, 2012
DOI:10.1021/mz300440q
One-dimensional nanostructures are self-assembled from an amphiphilic arylene-ethynylene macrocycle (AEM) in solution phase. The morphology and size of the nanostructures are controlled by simply changing the temperature, reversibly switching between monomolecular cross-sectioned nanofibers and large bundles. At elevated temperature in aqueous solutions, the tri(ethylene glycol) (Tg) side chains of the AEM become effectively more hydrophobic, thus facilitating intermolecular association through side chain interactions. The enhanced intermolecular association causes the ultrathin nanofibers to be bundled, forming an opaque dispersion in solution. The reported observation provides a simple molecular design rule that may be applicable to other macrocycle molecules for use in temperature-controlled assembly regarding both size and morphology.
Co-reporter:Aaron P. Esser-Kahn;Piyush R. Thakre;Hefei Dong;Jason F. Patrick;Vitalii K. Vlasko-Vlasov;Nancy R. Sottos;Scott R. White
Advanced Materials 2011 Volume 23( Issue 32) pp:
Publication Date(Web):
DOI:10.1002/adma.201190127
Co-reporter:Aaron P. Esser-Kahn;Piyush R. Thakre;Hefei Dong;Jason F. Patrick;Vitalii K. Vlasko-Vlasov;Nancy R. Sottos;Scott R. White
Advanced Materials 2011 Volume 23( Issue 32) pp:3654-3658
Publication Date(Web):
DOI:10.1002/adma.201100933
Co-reporter:Aaron D. Finke ; Dustin E. Gross ; Amy Han
Journal of the American Chemical Society 2011 Volume 133(Issue 35) pp:14063-14070
Publication Date(Web):July 20, 2011
DOI:10.1021/ja204795q
We present a crystallographic study that systematically investigates the effects of the n-alkyl side-chain length on the crystal packing in shape-persistent macrocycles. The solid-state packing of carbazole–ethynylene-containing macrocycles is sensitive to the alkyl-chain length. In macrocycles containing n-alkyl side chains up to nine carbons in length, face-on aromatic π interactions predominate, while the addition of one carbon leads to a completely different packing arrangement. Macrocycles with C10 or C11 chains exhibit a novel packing motif wherein the alkyl chains intercalate between macrocycles, leading in one case to continuous solvent-filled channels. When crystals of the C10 macrocycle are bathed in solvent, the included molecules exchange with the external solvent, and the alkyl chain disorder changes in response to changes in the guest volume in order to retain crystallinity. Powder X-ray diffraction data indicate that alkyl–macrocycle interactions in the longer chains “emulate” the distances typical of face-to-face π interactions, leading to deceptive indicators of π stacking.
Co-reporter:Matthew J. Kryger ; Alexander M. Munaretto
Journal of the American Chemical Society 2011 Volume 133(Issue 46) pp:18992-18998
Publication Date(Web):October 27, 2011
DOI:10.1021/ja2086728
Ultrasound activation of mechanophores embedded in polymer backbones has been extensively studied of late as a method for realizing chemical reactions using force. To date, however, there have been few attempts at systematically investigating the effects of mechanophore structure upon rates of activation by an acoustic field. Herein, we develop a method for comparing the relative reactivities of various cyclobutane mechanophores. Through the synthesis and ultrasonic irradiation of a molecular weight series of poly(methyl acrylate) polymers in which each macromolecule has a single chain-centered mechanophore, we find measurable and statistically significant shifts in molecular weight thresholds for mechanochemical activation that depend on the structure of the mechanophore. We also show that calculations based on the constrained geometries simulate external force method reliably predict the trends in mechanophore reactivity. These straightforward calculations and the experimental methods described herein may be useful in guiding the design and the development of new mechanophores for targeted applications.
Co-reporter:Koushik Ghosh
Journal of the American Chemical Society 2011 Volume 133(Issue 49) pp:19650-19652
Publication Date(Web):November 15, 2011
DOI:10.1021/ja2087163
We used fluorescence and electronic absorption spectroscopy to study the molecular weight dependence of macromolecule-induced folding in a chain-centered meta-phenylene ethynylene (mPE) oligomer. Analogous to the ability of intrinsically unstructured proteins (IUPs) to induce folding of globular proteins in cellular environments, we show that macromolecules attached to both ends of an mPE dodecamer induce the foldamer to collapse into a presumed helical conformation. The collapse is especially prominent once the macromolecule segments become larger than ca. 50 kDa. For sufficiently large macromolecules, the conformational structuring occurs even in solvents that normally denature the foldamer. Based on these findings, chain-centered foldamers might find use as models to investigate the fundamental macromolecular physics of IUPs.
Co-reporter:Erin L. Elliott, C. Scott Hartley and Jeffrey S. Moore
Chemical Communications 2011 vol. 47(Issue 17) pp:5028-5030
Publication Date(Web):28 Mar 2011
DOI:10.1039/C1CC11242B
Scrambling experiments suggest that the self-assembly of 2D ladders viaimine metathesis is kinetically trapped at four or more rungs. Consequently, ladders containing five or more rungs cannot be synthesized in high yield under the conditions used, as misaligned out-of-register byproducts cannot self-correct.
Co-reporter:Gerald O. Wilson, Mary M. Caruso, Stuart R. Schelkopf, Nancy R. Sottos, Scott R. White, and Jeffrey S. Moore
ACS Applied Materials & Interfaces 2011 Volume 3(Issue 8) pp:3072
Publication Date(Web):July 5, 2011
DOI:10.1021/am200584z
Dimethylnorbornene ester (DNE) is successfully used as a noncovalent adhesion promoter. DNE was confirmed to copolymerize with dicyclopentadiene (DCPD) to yield a copolymer with better adhesion to an EPON 828 epoxy matrix relative to poly(DCPD) alone. The mechanical properties of the copolymer were comparable to that of poly(DCPD) alone. An optimized blend of the monomers was encapsulated using a urea-formaldheyde microencapsulation procedure and the resulting capsules were used for in situ self-healing experiments. Improved healing efficiency was observed for samples containing the DCPD/DNE capsules under conditions in which the monomers were efficiently polymerized.Keywords: adhesion; fracture testing; microencapsulation; noncovalent interactions; ring-opening metathesis polymerization; self-healing polymers;
Co-reporter:Susan A. Odom, Aaron C. Jackson, Alex M. Prokup, Sarut Chayanupatkul, Nancy R. Sottos, Scott R. White, and Jeffrey S. Moore
ACS Applied Materials & Interfaces 2011 Volume 3(Issue 12) pp:4547
Publication Date(Web):November 24, 2011
DOI:10.1021/am201048a
We report a new core–shell microcapsule system for the visual detection of mechanical damage. The core material, 1,3,5,7-cyclooctatetraene, is a conjugated cyclic olefin and a precursor to intensely colored polyacetylene. A combination of poly(urea-formaldehyde) and polyurethane is required to effectively encapsulate the volatile core material. Increasing the outer shell wall thickness and including a core-side prepolymer improves the thermal stability and free-flowing nature of these capsules, which tend to leach and rupture with thinner shell walls. Capsules ruptured in the presence of the Grubbs–Love ruthenium catalyst show immediate color change from nearly colorless to red-orange and dark purple over time, and color change in thin films resulted from scratch damage.Keywords: color change; damage detection; encapsulation; microcapsule; ring-opening metathesis polymerization;
Co-reporter:Dustin E. Gross and Jeffrey S. Moore
Macromolecules 2011 Volume 44(Issue 10) pp:3685-3687
Publication Date(Web):April 21, 2011
DOI:10.1021/ma2006552
Co-reporter:Aaron P. Esser-Kahn, Susan A. Odom, Nancy R. Sottos, Scott R. White, and Jeffrey S. Moore
Macromolecules 2011 Volume 44(Issue 14) pp:5539-5553
Publication Date(Web):July 6, 2011
DOI:10.1021/ma201014n
Stimuli-responsive capsules are of interest in drug delivery, fragrance release, food preservation, and self-healing materials. Many methods are used to trigger the release of encapsulated contents. Here we highlight mechanisms for the controlled release of encapsulated cargo that utilize chemical reactions occurring in solid polymeric shell walls. Triggering mechanisms responsible for covalent bond cleavage that result in the release of capsule contents include chemical, biological, light, thermal, magnetic, and electrical stimuli. We present methods for encapsulation and release, triggering methods, and mechanisms and conclude with our opinions on interesting obstacles for chemically induced activation with relevance for controlled release.
Co-reporter:Aaron P. Esser-Kahn ; Nancy R. Sottos ; Scott R. White
Journal of the American Chemical Society 2010 Volume 132(Issue 30) pp:10266-10268
Publication Date(Web):July 13, 2010
DOI:10.1021/ja104812p
For the autonomous repair of damaged materials, microcapsules are needed that release their contents in response to a variety of physical and chemical phenomena, not just by direct mechanical rupture. Herein we report a general route to programmable microcapsules. This method creates core−shell microcapsules with polymeric shell walls composed of self-immolative polymer networks. The polymers in these networks undergo a head-to-tail depolymerization upon removal of the triggering end group, leading to breakdown of the shell wall and subsequent release of the capsule’s liquid interior. We report microcapsules with shell walls bearing both Boc and Fmoc triggering groups. The capsules release their contents only under conditions known to remove these triggering groups; otherwise, they retain their contents under a variety of conditions. In support of the proposed release mechanism, the capsule shell walls were observed to undergo physical cracking upon exposure to the triggering conditions.
Co-reporter:Matthew J. Kryger ; Mitchell T. Ong ; Susan A. Odom ; Nancy R. Sottos ; Scott R. White ; Todd J. Martinez
Journal of the American Chemical Society 2010 Volume 132(Issue 13) pp:4558-4559
Publication Date(Web):March 17, 2010
DOI:10.1021/ja1008932
Mechanical damage of polymers is often a destructive and irreversible process. However, desirable outcomes may be achieved by controlling the location of chain cleavage events through careful design and incorporation of mechanically active chemical moieties known as mechanophores. It is possible that mechanophores can be used to generate reactive intermediates that can autopolymerize or cross-link, thus healing mechanically induced damage. Herein we report the generation of reactive cyanoacrylate units from a dicyanocyclobutane mechanophore located near the center of a polymer chain. Because cyanoacrylates (which are used as monomers in the preparation of superglue) autopolymerize, the generated cyanoacrylate-terminated polymers may be useful in self-healing polymers. Sonication studies of polymers with the mechanophore incorporated into the chain center have shown that selective cleavage of the mechanophore occurs. Trapping experiments with an amine-based chromophore support cyanoacrylate formation. Additionally, computational studies of small-molecule models predict that force-induced bond cleavage should occur with greater selectivity for the dicyanocyclobutane mechanophore than for a control molecule.
Co-reporter:Zengxing Zhang ; Yanke Che ; Ronald A. Smaldone ; Miao Xu ; Benjamin R. Bunes ; Jeffrey S. Moore ;Ling Zang
Journal of the American Chemical Society 2010 Volume 132(Issue 40) pp:14113-14117
Publication Date(Web):September 17, 2010
DOI:10.1021/ja104105n
Foldamers are synthetic and designable oligomers that adopt a conformationally ordered state in selected solvents. We found that oligo(m-phenylene ethynylene)s, which are single-stranded foldamers, can be made to reversibly disperse and release single-walled carbon nanotubes (SWCNTs) simply by changing the solvent, consistent with a change from an unfolded state to a folded state. Using absorption spectroscopy, atomic force microscopy, Raman spectroscopy, and electrical measurements, we observed that the foldamer-dispersed SWCNTs are individually well-dispersed and have a strong interfacial interaction with the foldamers. In contrast, the released SWCNTs appeared to be free of foldamers. Under illumination, transistors based on the foldamer-dispersed SWCNTs demonstrated significant photoresponse, apparently due to photoinduced charge transfer between the foldamers and SWCNTs. The reported nanocomposites may open an alternative way of developing optoelectronic devices or sensors based on carbon nanotubes.
Co-reporter:Aaron D. Finke and Jeffrey S. Moore
Chemical Communications 2010 vol. 46(Issue 42) pp:7939-7941
Publication Date(Web):20 Sep 2010
DOI:10.1039/C0CC03113E
The substantial kinetic barrier to molybdenum nitride–alkyne metathesis is facilitated by precomplexation of the borane Lewis acid B(C6F5)3, providing convenient access to metathesis-active molybdenum alkylidynes. Spectroscopic and X-ray structural analysis suggest MoN bond weakening upon borane complexation.
Co-reporter:Ronald A. Smaldone;En-chi Lin
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 4) pp:927-935
Publication Date(Web):
DOI:10.1002/pola.23848
Abstract
A series of m-phenylene ethynylene (mPE) foldamers were crosslinked in their helical conformation using a reductive amination-based strategy. This was accomplished by placing aldehyde moieties in the backbone of the oligomer at specific residues, which allowed a diamine crosslinker to covalently link the helical loops together. Three different foldamers with crosslinks placed at different locations in the backbone were synthesized and characterized by mass spectrometry, 1H NMR, and gel permeation chromatography. The effect of the crosslinking on the stability of the folded state was evaluated through solvent denaturation studies. These studies show a reduction in the oligomer's ability to unfold of up to 30% relative to an unmodified mPE oligomer of the same length in solvents that promote unfolding. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 927–935, 2010
Co-reporter:Gerald O. Wilson;James W. Henderson;Mary M. Caruso;Benjamin J. Blaiszik;Patrick J. McIntire;Nancy R. Sottos;Scott R. White
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 12) pp:2698-2708
Publication Date(Web):
DOI:10.1002/pola.24053
Abstract
The suitability of various peroxide initiators for a radical polymerization-based self-healing system is evaluated. The initiators are compared using previously established criteria in the design of ring opening metathesis polymerization-based self-healing systems. Benzoyl peroxide (BPO) emerges as the best performing initiator across the range of evaluation criteria. Epoxy vinyl ester resin samples prepared with microcapsules containing BPO exhibited upwards of 80% healing efficiency in preliminary tests in which a mixture of acrylic monomers and tertiary amine activator was injected into the crack plane of the sample after the initial fracture. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 2698–2708, 2010
Co-reporter:Zheng Xue, Aaron D. Finke, and Jeffrey S. Moore
Macromolecules 2010 Volume 43(Issue 22) pp:9277-9282
Publication Date(Web):November 1, 2010
DOI:10.1021/ma102023a
An efficient synthesis of hyperbranched poly(m-phenylene)s is achieved by Suzuki polycondensation of a m-terphenyl-derived branched AB2 monomer. The resulting hyperbranched polymers show high molecular weight and relatively low polydispersity and are readily soluble in common organic solvents. A catalyst system composed of Pd(OAc)2 and S-Phos was found to be highly active in the present Suzuki polycondensation protocol. The molecular weight and polydispersity of the polymer are controllable by varying the catalyst loading and the starting monomer concentration. Experimental results are consistent with a pseudo-chain-growth pathway that involves intramolecular catalyst transfer during polymerization. Furthermore, the triflate end groups of the hyperbranched poly(m-phenylene)s can be efficiently converted to other functionalities via in situ Suzuki−Miyaura cross-couplings.
Co-reporter:Mary M. Caruso, Douglas A. Davis, Qilong Shen, Susan A. Odom, Nancy R. Sottos, Scott R. White and Jeffrey S. Moore
Chemical Reviews 2009 Volume 109(Issue 11) pp:5755
Publication Date(Web):October 14, 2009
DOI:10.1021/cr9001353
Co-reporter:Mary M. Caruso, Stuart R. Schelkopf, Aaron C. Jackson, Alexandra M. Landry, Paul V. Braun and Jeffrey S. Moore
Journal of Materials Chemistry A 2009 vol. 19(Issue 34) pp:6093-6096
Publication Date(Web):24 Jul 2009
DOI:10.1039/B910673A
Single-walled nanotubes (SWNTs) suspended in chlorobenzene (PhCl) and ethyl phenylacetate (EPA) were incorporated into microcapsules using an in situemulsification polymerization of urea-formaldehyde; the capsules release the SWNTs upon mechanical rupture.
Co-reporter:GeraldO. Wilson;KeithA. Porter;Haim Weissman;ScottR. White;NancyR. Sottos;JeffreyS. Moore
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 11-12) pp:1817-1825
Publication Date(Web):
DOI:10.1002/adsc.200900134
Abstract
The stability of second generation Grubbs’ alkylidenes to primary amines relative to the first generation derivatives is investigated. For both Grubbs’ alkylidene derivatives, the tricyclohexylphosphine (PCy3) ligand is displaced by n-butylamine and diethylenetriamine. However, while displacement of PCy3 in first generation Grubbs’ alkylidene derivatives results in decomposition of the catalyst, the N-heterocyclic carbene (NHC) ligand in second generation derivatives is not displaced by primary amines present in up to 100 equivalents. The result is the formation of new stable ruthenium-amine complexes. These complexes are characterized and their catalytic activity is evaluated in ring-closing metathesis (RCM) and ring-opening metathesis (ROMP) reactions. While the amine complexes evaluated were minimally active in RCM reactions, the ruthenium-butylamine complex was significantly active in ROMP and exhibited an initiation rate constant that was at least an order of magnitude greater than that of the second generation Grubbs’ alkylidene from which it was synthesized.
Co-reporter:Yunyi Lu, Jeffrey S. Moore
Tetrahedron Letters 2009 50(28) pp: 4071-4077
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.04.103
Co-reporter:Aaron D. Finke, Eric C. Elleby, Michael J. Boyd, Haim Weissman and Jeffrey S. Moore
The Journal of Organic Chemistry 2009 Volume 74(Issue 22) pp:8897-8900
Publication Date(Web):October 27, 2009
DOI:10.1021/jo902015w
Substoichiometric amounts of ZnCl2 promote the room temperature, Pd/P(t-Bu)3-catalyzed cross-coupling of aryl bromides with alkynes. Pd(I) dimer 1 is demonstrated to be a particularly active precatalyst for this reaction. The reaction is general for a wide variety of aryl bromides.
Co-reporter:Gavvalapalli Nagarjuna ; Jingshu Hui ; Kevin J. Cheng ; Timothy Lichtenstein ; Mei Shen ; Jeffrey S. Moore ;Joaquín Rodríguez-López
Journal of the American Chemical Society () pp:
Publication Date(Web):October 17, 2014
DOI:10.1021/ja508482e
Enhancing the ionic conductivity across the electrolyte separator in nonaqueous redox flow batteries (NRFBs) is essential for improving their performance and enabling their widespread utilization. Separating redox-active species by size exclusion without greatly impeding the transport of supporting electrolyte is a potentially powerful alternative to the use of poorly performing ion-exchange membranes. However, this strategy has not been explored possibly due to the lack of suitable redox-active species that are easily varied in size, remain highly soluble, and exhibit good electrochemical properties. Here we report the synthesis, electrochemical characterization, and transport properties of redox-active poly(vinylbenzyl ethylviologen) (RAPs) with molecular weights between 21 and 318 kDa. The RAPs reported here show very good solubility (up to at least 2.0 M) in acetonitrile and propylene carbonate. Ultramicroelectrode voltammetry reveals facile electron transfer with E1/2 ∼ −0.7 V vs Ag/Ag+(0.1 M) for the viologen 2+/+ reduction at concentrations as high as 1.0 M in acetonitrile. Controlled potential bulk electrolysis indicates that 94–99% of the nominal charge on different RAPs is accessible and that the electrolysis products are stable upon cycling. The dependence of the diffusion coefficient on molecular weight suggests the adequacy of the Stokes–Einstein formalism to describe RAPs. The size-selective transport properties of LiBF4 and RAPs across commercial off-the-shelf (COTS) separators such as Celgard 2400 and Celgard 2325 were tested. COTS porous separators show ca. 70 times higher selectivity for charge balancing ions (Li+BF4–) compared to high molecular weight RAPs. RAPs rejection across these separators showed a strong dependence on polymer molecular weight as well as the pore size; the rejection increased with both increasing polymer molecular weight and reduction in pore size. Significant rejection was observed even for rpoly/rpore (polymer solvodynamic size relative to pore size) values as low as 0.3. The high concentration attainable (>2.0 M) for RAPs in common nonaqueous battery solvents, their electrochemical and chemical reversibility, and their hindered transport across porous separators make them attractive materials for nonaqueous redox flow batteries based on the enabling concept of size-selectivity.
Co-reporter:Joshua A. Kaitz ; Charles E. Diesendruck
Journal of the American Chemical Society () pp:
Publication Date(Web):August 7, 2013
DOI:10.1021/ja405628g
End-capped poly(phthalaldehyde) (PPA) synthesized by anionic polymerization has garnered significant interest due to its ease of synthesis and rapid depolymerization. However, alternative ionic polymerizations to produce PPA have been largely unexplored. In this report, we demonstrate that a cationic polymerization of o-phthalaldehyde initiated by boron trifluoride results in cyclic PPA in high yield, with high molecular weight, and with extremely high cyclic purity. The cyclic structure is confirmed by NMR spectroscopy, MALDI-TOF mass spectrometry, and triple-detection GPC. The cyclic polymers are reversibly opened and closed under the polymerization conditions. Owing to PPA’s low ceiling temperature, cyclic PPA is capable of chain extension to larger molecular weights, controlled depolymerization to smaller molecular weights, or dynamic intermixing with other polymer chains, both cyclics and end-capped linears. These unusual properties endow the system with great flexibility in the synthesis and isolation of pure cyclic polymers of high molecular weight. Further, we speculate that the absence of end groups enhances the stability of cyclic PPA and makes it an attractive candidate for lithographic applications.
Co-reporter:Mary M. Caruso, Stuart R. Schelkopf, Aaron C. Jackson, Alexandra M. Landry, Paul V. Braun and Jeffrey S. Moore
Journal of Materials Chemistry A 2009 - vol. 19(Issue 34) pp:NaN6096-6096
Publication Date(Web):2009/07/24
DOI:10.1039/B910673A
Single-walled nanotubes (SWNTs) suspended in chlorobenzene (PhCl) and ethyl phenylacetate (EPA) were incorporated into microcapsules using an in situemulsification polymerization of urea-formaldehyde; the capsules release the SWNTs upon mechanical rupture.
Co-reporter:Erin L. Elliott, C. Scott Hartley and Jeffrey S. Moore
Chemical Communications 2011 - vol. 47(Issue 17) pp:NaN5030-5030
Publication Date(Web):2011/03/28
DOI:10.1039/C1CC11242B
Scrambling experiments suggest that the self-assembly of 2D ladders viaimine metathesis is kinetically trapped at four or more rungs. Consequently, ladders containing five or more rungs cannot be synthesized in high yield under the conditions used, as misaligned out-of-register byproducts cannot self-correct.
Co-reporter:Dustin E. Gross, Emre Discekici and Jeffrey S. Moore
Chemical Communications 2012 - vol. 48(Issue 37) pp:NaN4428-4428
Publication Date(Web):2012/03/20
DOI:10.1039/C2CC30701D
A dynamic combinatorial approach for the synthesis of arylene ethynylene macrocycles (AEMs) from linear polymers is described. By using readily available carbazolyl-ethynylene copolymers as starting materials we obtained a number of novel macrocycles that would be difficult to prepare by traditional methods.
Co-reporter:Aaron D. Finke and Jeffrey S. Moore
Chemical Communications 2010 - vol. 46(Issue 42) pp:NaN7941-7941
Publication Date(Web):2010/09/20
DOI:10.1039/C0CC03113E
The substantial kinetic barrier to molybdenum nitride–alkyne metathesis is facilitated by precomplexation of the borane Lewis acid B(C6F5)3, providing convenient access to metathesis-active molybdenum alkylidynes. Spectroscopic and X-ray structural analysis suggest MoN bond weakening upon borane complexation.
Co-reporter:Charles E. Diesendruck, Lingyang Zhu and Jeffrey S. Moore
Chemical Communications 2014 - vol. 50(Issue 87) pp:NaN13238-13238
Publication Date(Web):2014/07/30
DOI:10.1039/C4CC03514C
We investigated the use of mechanical stress to bend carbon–carbon triple bonds. Formation of an isoquinoline after reaction with a benzyl azide trap points towards a nucleophilic addition mechanism, differentiating mechanochemical trans-bending of π bonds from the typical reactivity observed for cisoidal bending of triple bonds in strained cyclic alkynes.
Co-reporter:Aniket Datar, Dustin E. Gross, Kaushik Balakrishnan, Xiaomei Yang, Jeffrey S. Moore and Ling Zang
Chemical Communications 2012 - vol. 48(Issue 71) pp:NaN8906-8906
Publication Date(Web):2012/07/16
DOI:10.1039/C2CC34127A
Large area uniform nanofibers have been fabricated from a hexameric arylene–ethynylene macrocycle (1) through in situ self-assembly on a glass substrate during solvent evaporation. The fibril morphology is controlled by the solvophilic core of 1, in conjunction with the interfacial interactions between the side chains of 1 and the substrate.
Co-reporter:Scott W. Sisco and Jeffrey S. Moore
Chemical Science (2010-Present) 2014 - vol. 5(Issue 1) pp:NaN85-85
Publication Date(Web):2013/09/02
DOI:10.1039/C3SC52018H
Chiral arylene–ethynylene macrocycles (AEMs) were synthesized via alkyne metathesis-mediated depolymerization of BINOL-based polymers. Homochiral dimers are selectively obtained from metathesis of heterochiral polymers. Thermodynamic analysis and computational modeling suggests the homochiral self-sorting to be entropy-driven due to the greater symmetry of the homochiral dimers over the heterochiral dimer. This symmetry-controlled reaction is a novel approach to achieving high selectivity in dynamic covalent macrocycle synthesis. Importantly, the result describes a new paradigm in dynamic covalent chemistry that will enable efficient synthesis of new chiral architectures.
Co-reporter:Preston A. May and Jeffrey S. Moore
Chemical Society Reviews 2013 - vol. 42(Issue 18) pp:NaN7506-7506
Publication Date(Web):2013/01/11
DOI:10.1039/C2CS35463B
Long chain polymers have a unique ability to become highly extended in elongational flow fields. The forces developed along the backbone give rise to scission of the chains near their center. Recently, this unique property of polymers has been adopted to explore new chemical transformations by embedding structural elements into the backbone designed to undergo site-specific bond cleavage, termed mechanophores. Experimental techniques to generate elongational flow fields exist in a variety of different arrangements and have been used to study polymer mechanochemistry in solution. This tutorial review will discuss progress in the field of polymer mechanochemistry as well as survey the techniques used to generate elongational flow fields. Ultrasonication will be highlighted as the technique that has been widely adopted to screen mechanophore reactivity in solution.

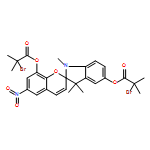
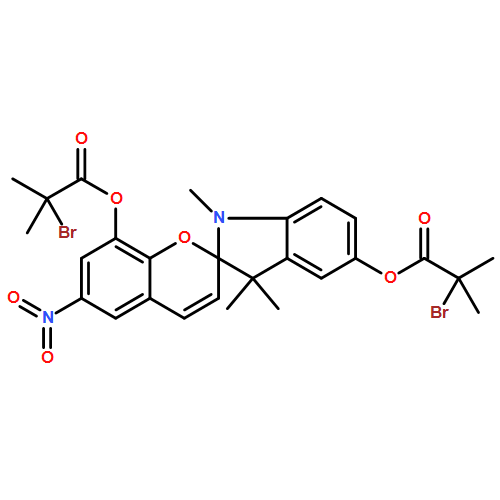
![2-Propenoic acid, 2-methyl-, 1,1'-(1',3'-dihydro-1',3',3'-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diyl) ester](/data/chemimg/3779800/1581285-94-0.png)
![2-Propenoic acid, 2-methyl-, 1,1'-(1',3'-dihydro-1',3',3'-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diyl) ester](/data/chemimg/3779800/1581285-94-0_b.png)










![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)














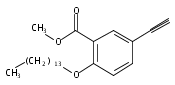
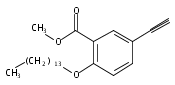




![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-1'(3'H)-ethanol, 5'-hydroxy-3',3'-dimethyl-6-nitro-](/data/chemimg/171100/1253909-23-7.png)
![Spiro[2H-1-benzopyran-2,2'-[2H]indole]-1'(3'H)-ethanol, 5'-hydroxy-3',3'-dimethyl-6-nitro-](/data/chemimg/171100/1253909-23-7_b.png)








![Phosphonic acid, [(3-iodophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/262000/261966-88-5.png)
![Phosphonic acid, [(3-iodophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/262000/261966-88-5_b.png)










![Tripropyl-[8-(tripropylazaniumyl)octyl]azanium;dibromide](http://img.cochemist.com/ccimg/106400/106327-24-6.png)
![Tripropyl-[8-(tripropylazaniumyl)octyl]azanium;dibromide](http://img.cochemist.com/ccimg/106400/106327-24-6_b.png)
![Tripropyl-[12-(tripropylazaniumyl)dodecyl]azanium;dibromide](http://img.cochemist.com/ccimg/106400/106327-23-5.png)
![Tripropyl-[12-(tripropylazaniumyl)dodecyl]azanium;dibromide](http://img.cochemist.com/ccimg/106400/106327-23-5_b.png)




![Phosphonic acid, [(2-iodophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/62700/62680-68-6.png)
![Phosphonic acid, [(2-iodophenyl)methyl]-, diethyl ester](http://img.cochemist.com/ccimg/62700/62680-68-6_b.png)
![3-[4-(HYDROXYMETHYL)PHENYL]-1-PROPANOL](http://img.cochemist.com/ccimg/36400/36308-36-8.png)
![3-[4-(HYDROXYMETHYL)PHENYL]-1-PROPANOL](http://img.cochemist.com/ccimg/36400/36308-36-8_b.png)





![Phenol, 4-[(phenylimino)methyl]-, 1-acetate](/data/chemimg/1079300/13031-46-4.png)
![Phenol, 4-[(phenylimino)methyl]-, 1-acetate](/data/chemimg/1079300/13031-46-4_b.png)













![Propanoic acid, 2-bromo-2-methyl-, 2-[5'-(2-bromo-2-methyl-1-oxopropoxy)-3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'(3'H)-yl]ethyl](/data/chemimg/3779200/1585963-26-3.png)
![Propanoic acid, 2-bromo-2-methyl-, 2-[5'-(2-bromo-2-methyl-1-oxopropoxy)-3',3'-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indol]-1'(3'H)-yl]ethyl](/data/chemimg/3779200/1585963-26-3_b.png)
![2-Propenoic acid, 2-methyl-, 1,1'-(1',3'-dihydro-1',3',3'-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diyl) ester, polymer with 1,1'-(1,2-](/data/chemimg/3779800/1581285-95-1.png)
![2-Propenoic acid, 2-methyl-, 1,1'-(1',3'-dihydro-1',3',3'-trimethyl-6-nitrospiro[2H-1-benzopyran-2,2'-[2H]indole]-5',8-diyl) ester, polymer with 1,1'-(1,2-](/data/chemimg/3779800/1581285-95-1_b.png)





![Poly[(1,3-dihydro-1,3-isobenzofurandiyl)oxy]](http://img.cochemist.com/ccimg/27000/26966-00-7.png)
![Poly[(1,3-dihydro-1,3-isobenzofurandiyl)oxy]](http://img.cochemist.com/ccimg/27000/26966-00-7_b.png)