Co-reporter:Xiaolei Zhang, Hongning Zheng, Jie Li, Fei Xu, Jing Zhao, and Hong Yan
Journal of the American Chemical Society October 18, 2017 Volume 139(Issue 41) pp:14511-14511
Publication Date(Web):August 6, 2017
DOI:10.1021/jacs.7b07160
Carboranyl aldehydes are among the most useful synthons in derivatization of carboranes. However, compared to the utilization of carboranyl carboxylic acids in selective B–H bond functionalizations, the synthetic application of carboranyl aldehydes is limited due to the weakly coordinating nature of the aldehyde group. Herein, the direct arylation of o-carboranyl aldehydes has been developed via Pd-catalyzed cage B–H bond functionalization. With the help of glycine to generate a directing group (DG) in situ, a series of cage B(4,5)-diarylated- and B(4)-monoarylated-o-carboranyl aldehydes were obtained in good to excellent yields with high selectivity. A wide range of functional groups are tolerated. The aldehyde group in the B–H arylated products could be readily removed or transformed into o-carboranyl methanol. A plausible catalytic cycle for B–H arylation was proposed based on control experiments and stoichiometric reactions, including the isolation of a key bicyclic palladium complex.
Co-reporter:Xiang Li;Yongheng Yin;Changsheng Lu;Qiang Zhao
Dalton Transactions 2017 vol. 46(Issue 30) pp:10082-10089
Publication Date(Web):2017/08/01
DOI:10.1039/C7DT02009K
A novel series of heteroleptic iridium complexes with 2-phenyl-pyridine as a main ligand and carborane-functionalized 2,2′-bipyridine as an ancillary ligand were synthesized, and characterized as [Ir(ppy)2(By)]PF6 (where ppy is 2-phenyl-pyridine, By is 5-(2-R-Cb)-2,2′-bipyridine, R = H (2a), CH3 (2b), Ph (2c), iPr (2d) and iBu (2e), or By is 4-(2-R-Cb)-2,2′-bipyridine while R = H (3a), CH3 (3b), Ph (3c), iPr (3d) and iBu (3e), Cb = o-carboran-1-yl). The R groups and the substitution sites of carborane on the pyridine ring have caused differences in the emission properties of these complexes. In addition, the quantum efficiency of [Ir(ppy)2(By)]PF6 complexes has been tuned as well through the introduction of various 2-R-substituted o-carboranes into the ancillary ligand 2,2′-bipyridine, no matter in the solid state (from 0.12 to 0.25) or in solution (from 0.04 to 0.25). The emission color was tuned from yellow to red by the o-carboranyl unit because of its inductive effect. Density functional theory (DFT) and time dependent DFT (TD-DFT) calculations have been applied to investigate excited-state electronic structures of the newly synthesized complexes, which are consistent with the observed red-shift emissions.
Co-reporter:Xiang Li;Yongheng Yin;Pengli Gao;Weijie Li;Changsheng Lu;Qiang Zhao
Dalton Transactions 2017 vol. 46(Issue 40) pp:13802-13810
Publication Date(Web):2017/10/17
DOI:10.1039/C7DT03097E
The structure–property relationship of carborane-embedded cationic iridium(III) complexes was investigated in this work, especially with donor–acceptor-type ligands. Firstly, an efficient synthetic approach for the new donor–acceptor-type ligands (2 and 4) was developed. By using these ligands, novel iridium(III) complexes II and IV were prepared. Complex IV shows about a 92 nm red-shift in solution (ΦP = 0.13 in ethanol), and the emission color has an obvious change from green to red when compared with the Model complex. In addition, the photophysical characteristics of complex IV are quite sensitive to the oxygen level in living cells, based on which endocellular hypoxia imaging of complex IV has been realized by the phosphorescence lifetime imaging microscopy (PLIM) technology. Besides the experimental studies, density functional theory (DFT) and time dependent DFT (TD-DFT) calculations have been successfully applied to investigate the excited-state electronic structures of carborane-modified iridium complexes.
Co-reporter:Xiang Li;Xiao Tong;Yongheng Yin;Changsheng Lu;Wei Huang;Qiang Zhao
Chemical Science (2010-Present) 2017 vol. 8(Issue 9) pp:5930-5940
Publication Date(Web):2017/08/21
DOI:10.1039/C7SC00160F
Mitochondria as vital intracellular organelles play critical roles in multiple physiological processes, and their polarity is a crucial characteristic that can reveal the intracellular environment and impact cellular events. In this work, we designed and synthesized a novel series of highly emissive and environmentally sensitive phosphorescent iridium(III) complexes (2a–2e, 3a–3e and 4) functionalized by o-carborane. These complexes showed high emission quantum yields both in solution and in solid state (up to ΦPL = 0.82), long emission lifetime and tunable emission wavelength over 74 nm by introduction of a carboranyl motif in their ligands. Importantly, all the complexes have shown significant solvatochromic effects in contrast to the carborane-free control complex. Among them, complex 2d shows the highest sensitivity to polarity of solvents with a MPPS (maximum peak phosphorescence shift) value of 42 nm and clear dependence of phosphorescence lifetime on solvent polarity. Interestingly, complex 2d can easily penetrate into cells and preferentially distribute in mitochondria. To utilize these properties, the first phosphorescent imaging of mitochondrial polarity has been realized by photoluminescence lifetime imaging microscopy (PLIM), which can monitor mitochondria-relevant cellular processes such as cell apoptosis and distinguish cancer cells from normal cells. Compared to intensity-based sensing, lifetime-based detection is independent of the probe concentration, excitation power and photobleaching of probes, which can show high accuracy and reproducibility.
Co-reporter:Huan-huan Li, Fan Bai, Hong YanChang-sheng Lu
Organic Letters 2017 Volume 19(Issue 4) pp:
Publication Date(Web):February 1, 2017
DOI:10.1021/acs.orglett.7b00013
Base-promoted decarboxylative azo couplings of carboranyl carboxylic acids with diazo salts have been developed to provide trans-azocarboranes in high yields (up to 94%). This approach is simple, efficient, and compatible with various functional groups. Mechanistically, the coupling has been proven to proceed in a nonradical pathway, which is distinct from those classical decarboxylative couplings.
Co-reporter:Deshuang Tu;Pakkin Leong;Song Guo; Dr. Hong Yan;Changsheng Lu; Dr. Qiang Zhao
Angewandte Chemie 2017 Volume 129(Issue 38) pp:11528-11532
Publication Date(Web):2017/09/11
DOI:10.1002/ange.201703862
AbstractThe development of organic single-molecule solid-state white emitters holds a great promise for advanced lighting and display applications. Highly emissive single-molecule white emitters were achieved by the design and synthesis of a series of o-carborane-based luminophores. These luminophores are able to induce multiple emissions to directly emit high-purity white light in solid state. By tuning both molecular and aggregate structures, a significantly improved white-light efficiency has been realized (absolute quantum yield 67 %), which is the highest value among the known organic single-molecule white emitters in the solid state. The fine-tuning of the packing modes from H- to J- and cross-stacking aggregates as well as intermolecular hydrogen bonds are successful in one molecular skeleton. These are crucial for highly emissive white-light emission in the solid state.
Co-reporter:Huanhuan Li;Fan Bai;Changsheng Lu;Vladimir I. Bregadze
European Journal of Organic Chemistry 2017 Volume 2017(Issue 10) pp:1343-1352
Publication Date(Web):2017/03/10
DOI:10.1002/ejoc.201601537
An iridium-catalyzed cage B–H sulfonamidation of o-carborane directed by a carboxylic acid group is reported that proceeds in the absence of ligands or external oxidants. A series of sulfonyl azides can be selectively sulfonamidated at the B(4) site in high yields with excellent functional group tolerance. This approach can also be applied to aryl and aliphatic azides. Innocuous CO2 and N2 were released as byproducts. In addition, the carboxylic acid group can be easily removed under mild conditions.
Co-reporter:Xiaolei Zhang; Huimin Dai; Hong Yan; Wenli Zou;Dieter Cremer
Journal of the American Chemical Society 2016 Volume 138(Issue 13) pp:4334-4337
Publication Date(Web):February 24, 2016
DOI:10.1021/jacs.6b01249
For the first time, nonclassical hydrogen (H)-bonding involving a B–H···π interaction is described utilizing both quantum chemical predictions and experimental realization. In the gas phase, a B–H···π H-bond is observed in either B2H6···benzene (ΔE = −5.07 kcal/mol) or carborane···benzene (ΔE = −3.94 kcal/mol) complex at reduced temperatures. Ir-dimercapto-carborane complexes [Cp*Ir(S2C2B10H10)] are designed to react with phosphines PR3 (R = C6H4X, X = H, F, OMe) to give [Cp*Ir(PR3)S2C2B10H10] for an investigation of B–H···π interactions at ambient temperatures. X-ray diffraction studies reveal that the interaction between the carborane BH bonds and the phosphine aryl substituents involves a BH···π H-bond (H···π distance: 2.40–2.76 Å). 1H NMR experiments reveal that B–H···π interactions exist in solution according to measured 1H{11B} signals at ambient temperatures in the range 0.0 ≤ δ ≤ 0.3 ppm. These are high-field shifted by more than 1.5 ppm relative to the 1H{11B} signals obtained for the PMe3 analog without B–H···π bonding. Quantum chemical calculations suggest that the interaction is electrostatic and the local (B)H···ring stretching force constant is as large as the H-bond stretching force constant in the water dimer.
Co-reporter:Huimin Dai, Chao Yu, Zihao Wang, Hong Yan, and Changsheng Lu
Organic Letters 2016 Volume 18(Issue 14) pp:3410-3413
Publication Date(Web):June 28, 2016
DOI:10.1021/acs.orglett.6b01566
The Heck reaction between arenes and allyl acetate has led to cinnamyl derivatives and allyl products depending on the regioselectivity of β-elimination. The regioselectivity can be controlled by the solvent in the Rh(III)-catalyzed arene–allyl acetate coupling via C–H activation: (1) in THF, cinnamyl derivatives via β-H elimination were generated; (2) in MeOH, allyl products via β-OAc elimination were produced. Both routes have advantages such as excellent γ-selectivity toward allyl acetate, good to excellent yields, and broad substrate scope.
Co-reporter:Chun-Xiao Li, De-Shuang Tu, Rui Yao, Hong Yan, and Chang-Sheng Lu
Organic Letters 2016 Volume 18(Issue 19) pp:4928-4931
Publication Date(Web):September 28, 2016
DOI:10.1021/acs.orglett.6b02413
An effective photoredox-mediated tandem phosphorylation/cyclization reaction of diphenylphosphine oxide with three types of radical acceptors leads to P(O)Ph2-containing phenanthridines, isoquinolines, and indolin-2-ones by formation of both C–P and C–C bonds. [Ir(ppy)2(dtbpy)]PF6 (1 mol %) was used as the catalyst, CsF or Cs2CO3 as the base, and K2S2O8 as the oxidant. A series of functional groups can be tolerated at room temperature. Moderate to good yields were generated.
Co-reporter:Huimin Dai;Chao Yu;Changsheng Lu
European Journal of Organic Chemistry 2016 Volume 2016( Issue 7) pp:1255-1259
Publication Date(Web):
DOI:10.1002/ejoc.201501551
Abstract
A RhIII-catalyzed C–H allylation of electron-deficient arenes, heteroarenes, and alkenes at room temperature was developed with allyl bromide. The reaction was carried out in diethyl ether without dehydration, and C–H activation was assisted by the directing anionic nitrogen of the aniline-derived amide. Following the allylation, a domino cycling synthesis of 3,4-dihydroisoquinolin-1(2H)-ones with N-bromosuccinimide (NBS) through intramolecular aminobromination of the introduced double bond was achieved.
Co-reporter:Huimin Dai, Guifeng Liu, Xiaolei Zhang, Hong Yan, and Changsheng Lu
Organometallics 2016 Volume 35(Issue 10) pp:1488-1496
Publication Date(Web):March 15, 2016
DOI:10.1021/acs.organomet.6b00082
The reactions of the 16e half-sandwich complex CpCoS2C2B10H10 (1), diazo esters, and various 1,6-diynes (3a–i; PhN(CH2C≡CH)2, 4-Me-PhN(CH2C≡CH)2, 4-OMe-PhN(CH2C≡CH)2, 4-F-PhN(CH2C≡CH)2, BzN(CH2C≡CH)2, O(CH2C≡CH)2, C(Ac)2(CH2C≡CH)2, N(CH2C≡CH)3, NH(CH2C≡CH)N(CH2C≡CH)2) were investigated, in which two novel types of B–H activated products CpCoS2B10H9(CH2CO2Et)C5H3N(R)(CH═CHCO2Et) (4a–c; R = Ph, 4-Me-Ph, 4-OMe-Ph) and the key intermediate CpCoS2B10H9(CHCO2Me) (CH2CO2Me) (9) were isolated. 9 features a reactive Co–B bond, which triggers insertion of various 1,6-diynes to further lead to different final products. Substrates 3a–c are activated by the Co–B bond to produce o-carborane derivatives 4a–c which are functionalized by a cobalt-complexed η3-pyrrolylmethyl group. The pyrrole ring is formed by in situ ring closure of 1,6-diynes. Control experiments and isolation of the intermediate CpCoS2B10H9(CHCO2Me)(CH2CO2Me)HC═CCH2N(4-Me-Ph)(CH2C≡CH) (10) support the proposed mechanism concerning the formation of 4a–c analogues by oxidation. All of the new complexes were characterized by NMR, IR, elemental analysis, and mass spectrometry. The structures of 4a–6a and 9 were determined by single-crystal X-ray diffraction analysis as well.
Co-reporter:Xiang Li;Dr. Hong Yan;Dr. Qiang Zhao
Chemistry - A European Journal 2016 Volume 22( Issue 6) pp:1888-1898
Publication Date(Web):
DOI:10.1002/chem.201503456
Abstract
Phosphorescent transition-metal complexes (PTMCs) have attracted great interest because of their excellent properties which may lead to promising applications in optoelectronics. In recent years, carboranes have been demonstrated to be a novel and effective tool to tune phosphorescence of PTMCs. This Concept article deals with the advances of carborane-functionalized PTMCs for potential optical applications. The discussions are focused on the design strategies and synthetic procedures leading to carborane-functionalized PTMCs, the influence of carboranes on the optical properties of PTMCs, and the promising optical applications of this interesting class of phosphorescent materials.
Translation abstract
Co-reporter:Changyu Wu, Afzal Shah, Hongde Ye, Xiao Chen, Jing Ye, Hui Jiang, Baoan Chen, Xuemei Wang, Hong Yan
Analytica Chimica Acta 2015 Volume 857() pp:39-45
Publication Date(Web):1 February 2015
DOI:10.1016/j.aca.2014.12.019
•Electrochemical behaviors of novel ferrocenyl based carboranes (FcCB) were explored with a droplet system.•The shifts of peak potentials with changes of pH values indicated the involvement of proton during electron transfer reaction.•Normal cells and cancer cells could be specifically recognized by using FcCB as probe.•This electrochemical method in a droplet shows great potential application for relevant diagnostics of clinical samples.Novel ferrocenyl based carboranes (FcCBs) and their distinguish behavior for cancer cell recognition have been explored in this contribution. The voltammetric study in a droplet of 10 μL placed on the surface of a glassy carbon electrode demonstrates the excellent electrochemical behavior of FcCBs, which could be further exploited for establishing the promising and sensitive biosensors. The FcCBs’ redox behavior is examined in a wide pH range, and square wave voltammetry revealed the reversible and irreversible nature of first and second anodic peaks. The obvious shifts in peak potentials corresponding with the change of pH values demonstrate the abstraction of electrons to be accompanied with the transfer of protons. By using the droplet electrochemical technique, FcCBs can be employed to distinguish normal and cancer cells with a linear range from 1.0 × 103 to 3.0 × 104 cells mL−1 and the limit of detection at 800 cells mL−1. The novel carborane derivatives could be utilized as important potential molecular probes for specific recognition of cancer cells like leukemia cells from normal cells.
Co-reporter:Yi-fei Ji;Qi-bai Jiang
European Journal of Organic Chemistry 2015 Volume 2015( Issue 9) pp:2051-2060
Publication Date(Web):
DOI:10.1002/ejoc.201403510
Abstract
Nitro compounds are important intermediates in synthetic organic chemistry and the chemical industry. Herein, the efficient copper-catalyzed [10 % Cu(NO3)2·3H2O] nitration of anilides was developed by using TBN (tert-butyl nitrite) as a nitrating reagent to give the corresponding nitro-substituted aromatic products in good to excellent yields. The use of TBN also led to the selective nitration of acrylamides at room temperature to afford only the (E) isomer of the nitration product. A series of anilides and acrylamides with a broad array of functional groups were well-tolerated by this procedure. This synthetic method has many advantages, which include inexpensive starting materials, mild reaction conditions, a fast reaction rate, and high yields. A mechanistic investigation indicates that a nitro radical, which is generated from the thermal homolysis of TBN, is involved in the two nitration processes.
Co-reporter:Guifeng Liu and Hong Yan
Organometallics 2015 Volume 34(Issue 3) pp:591-598
Publication Date(Web):January 16, 2015
DOI:10.1021/om501016w
The three-component reactions of the 16-electron half-sandwich complex CpCo(S2C2B10H10) (Cp = cyclopentadienyl) (1) with ethyl diazoacetate (EDA) and alkynes R1≡R2 (R1 = Ph, R2 = H; R1 = CO2Me, R2 = H; R1 = R2 = CO2Me; R1 = Fc, R2 = H) at ambient temperature lead to compounds CpCo(S2C2B10H9)(CH2CO2Et) (CHCO2Et)(R1═R2) (2–5), CpCo(S2C2B10H9)(CH2CO2Et)(R2–R1–CHCO2Et) (6–9), CpCo(S2C2B10H9)(CH2CO2Et)(CH(Ph)C═CHCO2Et) (10), and CpCo(S2C2B10H9)(CH2CO2Et)(CH(Fc)–CH–CCO2Et) (11). In 2–5, one alkyne is stereoselectively inserted into the Co–B bond, one EDA molecule is used to form a sulfide ylide, and the second EDA molecule is inserted into one Co–S bond to form a three-membered metallacyclic ring. At ambient temperature 2–5 undergo rearrangement to 6–9 through migratory insertion of the inserted EDA. Different from 2–5, in 10 phenylacetylene is inserted into the Co–B bond at the terminal carbon and the terminal carbon is coupled with one EDA to afford a six-membered metallacyclic ring with the C═O coordination to metal. In 11, a stable Co–B bond is generated, and one EDA and one ethynylferrocene are inserted into the Co–S bond. Moreover, if weakly basic silica is present, 2–4 can lose an apex BH close to the two carbon atoms of o-carborane to give rise to CpCo(S2C2B9H9)(CH2CO2Et)2(R1═R2) (12–14) accompanied by the coordination of the two sulfide ylide units to the metal center. The solid-state structures of 2–4, 6–12, and 14 were characterized by X-ray structural analysis.
Co-reporter:Lin Zhu;Xiao Tang;Qi Yu;Wen Lv; Hong Yan;Qiang Zhao;Wei Huang
Chemistry - A European Journal 2015 Volume 21( Issue 12) pp:4721-4730
Publication Date(Web):
DOI:10.1002/chem.201405897
Abstract
2-Thienylpyridyl iridium(III) complexes containing an o-, m-, or p-carboranylvinyl-2,2′-bipyridine ligand and various counteranions (denoted o-PF6, m-BF4, m-PF6, m-SbF6, m-ClO4, m-OTf, m-NO3, m-BPh4, m-F, m-Cl, and p-PF6) were synthesized by using C-formyl carboranes as starting materials. The solid-state structures of o-PF6, m-PF6, m-ClO4, and m-BF4 showed that the cations form twisted cavities in which the anions are fixed by multiple hydrogen bonds. Anion–hydrogen interactions were investigated for nine m-carborane-based complexes with different counteranions. All carborane-based iridium(III) complexes show similar phosphorescence yields in solution but significantly different emission in the solid state. Anion-exchange titration and theoretical calculations revealed the relationships between structures and optical properties. The size of the anion and CH⋅⋅⋅X anion–hydrogen bonds strongly influence the phosphorescence quantum yield in the solid state. In particular, the CcarH⋅⋅⋅X hydrogen bonds between the carboranyl unit and the anion play an important role in solid-state phosphorescence. Complex p-PF6 was successfully applied in phosphorescence-lifetime bioimaging owing to its low toxicity and near-infrared emission.
Co-reporter:Xiaolei Zhang, Zhiwen Zhou and Hong Yan
Chemical Communications 2014 vol. 50(Issue 86) pp:13077-13080
Publication Date(Web):05 Sep 2014
DOI:10.1039/C4CC06171C
New o-carborane-9,12-dithiolate dirhodium complexes are reported to selectively activate inert B–H bonds of o-carboranes to form B–X bonds by using water, alcohols, and alkylhalides (<60 °C, yields >80%). Characterization of the key intermediates demonstrates an oxidative addition and reductive elimination pathway via metal–metal cooperativity.
Co-reporter:Wei Zhong, Qibai Jiang, Qian Zhang, Yi Shang, Hong Yan and Vladimir Bregadze
Dalton Transactions 2014 vol. 43(Issue 13) pp:4962-4968
Publication Date(Web):24 Sep 2013
DOI:10.1039/C3DT52308J
Reactions of half-sandwich complex Cp*IrS2C2B10H10 (1) with 1-azido-3-nitrobenzene (3-NO2C6H4N3, L) upon heating or under light led to new complexes 2–6. Complexes 2 and 3 contain a five-membered cyclometalated ligand arising from C(sp2)–H activation of the azide ligand L. Complex 4 is a 16 electron species containing a new-generated C–C bond between the azide ligand L and the Cp* unit where C(sp3)–H activation of the methyl unit occurred. Complexes 5 and 6 contain two types of the ligand which appear in complexes 2, 3 and 4. Further reactions of complexes 5 and 6 with L under more harsh conditions gave rise to the nucleophilic addition products 7 and 8, where ring expansion of the azide ligand at the imido site of complexes 5 and 6 happened. Complexes 2–8 were characterized by NMR, MS, IR, and elemental analysis, and X-ray structural analyses were performed for complexes 2–4 and 6–8. The radical mechanisms for the formation of complexes 2–6 were proposed on the basis of capture experiments by EPR and ESI-MS. And the formation mechanism of complexes 7 and 8 was also suggested.
Co-reporter:Wei Zhong, Mingshi Xie, Yizhi Li and Hong Yan
RSC Advances 2014 vol. 4(Issue 106) pp:61799-61808
Publication Date(Web):31 Oct 2014
DOI:10.1039/C4RA13017K
A three-component reaction of the 16-electron half-sandwich complex Cp*Co(S2C2B10H10) (2) with methyl diazoacetate (MDA) and toluenesulphonyl azide (TsN3) gave di-inserted products Cp*Co(S2C2B10H10)(C–CO2Me)(CHCO2Me)(NHTs) (3) and Cp*Co(S2C2B10H10)(CHCO2Me)(CHCO2Me)(N3Ts) (4), where MDA and TsN3 in a molar ratio of 2:1 were inserted into the Co–S bond to form a five-membered metallacyclic ring with different coordination modes. Its two-component reaction with MDA affording complexes Cp*Co(S2C2B9H9)(CH2CO2Me)(CHCO2Me) (5), Cp*Co(S2C2B10H10)(CH2CO2) (6) and Cp*Co(S2C2B9H9) (CH2CO2) (7) proved that the abovementioned three-component reaction is not a simple stepwise reaction. For another 16-electron half-sandwich complex CpCo(S2C2B10H10) (1), two stepwise reaction routes were designed to achieve its incorporation with MDA and TsN3. The reaction occurred via the alkylidene-bridged adduct CpCo(S2C2B10H10)(CHCO2Me) (8) or the imido-bridged adduct CpCo(S2C2B10H10)(NTs) (9), and complex CpCo(S2C2B10H10)(CHCO2Me)(NTs) (10) was isolated as the sole product in moderate yield. In each reaction route, one molecule of MDA and one molecule of TsN3 insert into the two Co–S bonds of complex 1, respectively, with the loss of N2. All the new complexes were characterized using NMR spectroscopy, mass spectrometry, IR spectroscopy, elemental analysis and X-ray structural analysis.
Co-reporter:Chao Shi;Deshuang Tu;Qi Yu;Hua Liang;Yahong Liu;Zhihong Li;Dr. Hong Yan;Dr. Qiang Zhao;Dr. Wei Huang
Chemistry - A European Journal 2014 Volume 20( Issue 50) pp:16550-16557
Publication Date(Web):
DOI:10.1002/chem.201404743
Abstract
New iridium tetrazolate complexes containing o-, m-, or p-carboranyl substitution in different positions of a phenylpyridine ligand have been prepared. The carborane isomers and the effect of their substitution position in the tuning of optical properties have been examined. The neutral complexes with the carboranyl substituent on the phenyl ring in meta position relative to the metal exhibit redshifted emission bands in contrast to blueshifts for those with carboranyl in para position. All cationic complexes display evidently blueshifted dual-peak emission compared with the carborane-free complex (c-TZ) with a broad single-peak emission. Introduction of carborane leads to a blueshift over 70 nm relative to c-TZ. Carboranes also significantly improve phosphorescence efficiency (ΦP) and lifetime (τ), that is, ΦP=0.64 versus 0.21 (c-TZ) and τ=880 ns versus 241 ns (c-TZ). The unique hydrophilic nido-carborane-based IrIII complex nido-o-1 shows the largest phosphorescence efficiency (abs ΦP=0.57) among known water-soluble iridium complexes, long emission lifetime (τ=4.38 μs), as well as varying emission efficiency and lifetime with O2 content in aqueous solution. Therefore, nido-o-1 has been used as an excellent oxygen-sensitive phosphor for intracellular O2 sensing and hypoxia imaging.
Co-reporter:Xiaolei Zhang, Xiaodong Zou, and Hong Yan
Organometallics 2014 Volume 33(Issue 10) pp:2661-2666
Publication Date(Web):May 15, 2014
DOI:10.1021/om500411b
We present the first synthesis and characterization of a series of boron-fused 1,4-dithiin compounds through the reactions of newly established boron-substituted 16e half-sandwich complex Cp*Co(9,12-S2C2B10H10) with alkynes. The generated C2S2B2 ring in these 1,4-dithiin species is a stable structural motif with electron-negative sulfur atoms, as evidenced by theoretical calculation and its solid-state self-assembly. Single-crystal X-ray analysis indicates that Ccarb–H···S hydrogen bonding is involved in the self-assemblies of these compounds, which serves as a compatible interaction with stronger Ccarb–H···π, Ccarb–H···O, or Ccarb–H···F hydrogen bonding. All new compounds are characterized by NMR, mass spectroscopy, and X-ray structural analysis.
Co-reporter:Zhaojin Wang ; Hongde Ye ; Yuguang Li ; Yizhi Li
Journal of the American Chemical Society 2013 Volume 135(Issue 30) pp:11289-11298
Publication Date(Web):July 8, 2013
DOI:10.1021/ja4047075
The 16-electron complex CpCoS2C2B10H10 (1) is found to react with the alkynes HC≡CC(O)R [R = methyl (Me), phenyl (Ph), styryl (St), ferrocenyl (Fc)] at ambient temperature to give two types of 17-electron cobalt complexes 2a–d and 3a–d containing unique B(3)/B(6)-norbornyl carborane moieties. A formation mechanism via a tandem sequence of metal-induced B–H activation, B–Cp formation, Cp delivery and Diels–Alder addition is proposed on the basis of DFT calculations. The reactivity of these paramagnetic 17-electron complexes has been studied: Exposed to a combination of air, moisture and silica, complexes 2a–d undergo alkyl C–S cleavage to give 16-electron complexes 4a–c containing a boron-norbornadienyl moiety, and simultaneous carboranyl C–S cleavage to afford cobalt-free carborane derivatives 5a–d containing a boron-norbornyl unit. Both 2a–d and 3a–d allow further alkyne insertion into the Co–S bond to generate cobalt-free boron–norbornyl carborane derivatives (Z/E)-7a–d and (Z/E)-8a–d, both containing a vinyl sulfido group. Addition of AlCl3 not only promotes the conversion of 2a–d, but also leads predominantly to (E)-9a–d as retro-Diels–Alder products. Upon heating, the isomerization from E to Z-configuration of the vinyl group and reorganization of the norbornyl moiety of (Z/E)-7a–d occur to lead to (Z)-9a–d as well as the unexpected [1,2]-H shifted products (Z)-10b,c. Thus, the 17-electron complexes 2a–d and 3a–d serve as intermediates for synthesis of variety of boron-functionalized carborane derivatives. In this study, efficient routes have been developed through cobalt-mediated B–H activation to prepare boron-functionalized carborane derivatives that are unavailable by conventional routes.
Co-reporter:Lin Zhu, Wen Lv, Shujuan Liu, Hong Yan, Qiang Zhao and Wei Huang
Chemical Communications 2013 vol. 49(Issue 90) pp:10638-10640
Publication Date(Web):18 Sep 2013
DOI:10.1039/C3CC46276E
Carborane-incorporated (at both carbon and boron sites) tribranched fluorophores were efficiently prepared. oB significantly improves the fluorescence quantum yield. mC exhibits the largest two-photon absorption among the known carborane-based derivatives and has been successfully applied in two-photon fluorescence 2D & 3D bioimaging.
Co-reporter:Qibai Jiang, Zhaojin Wang, Yizhi Li and Hong Yan
Chemical Communications 2013 vol. 49(Issue 52) pp:5880-5882
Publication Date(Web):13 May 2013
DOI:10.1039/C3CC42554A
A 15-electron Ru(III) complex containing carborane-thiolato ligands heterolytically cleaves dihydrogen at ambient pressure and temperature to give Ru(IV)/Ru(II) species containing a hydrido ligand at Ru(IV) and SH ligand at Ru(II). In the presence of HBF4·H2O the reaction further leads to an unprecedented replacement of an S atom by an O atom in the carborane-dithiolato ligand.
Co-reporter:Chao Shi, Huibin Sun, Qibai Jiang, Qiang Zhao, Jingxia Wang, Wei Huang and Hong Yan
Chemical Communications 2013 vol. 49(Issue 42) pp:4746-4748
Publication Date(Web):04 Mar 2013
DOI:10.1039/C3CC40996A
Both neutral and cationic iridium(III) complexes containing carborane units were synthesized. Bulky carboranes can significantly improve phosphorescence quantum yields of these complexes and the electronic effect of carboranes can evidently tune emission wavelengths of cationic complexes.
Co-reporter:Xiao Tang, Zhaojin Wang, Yizhi Li, Hong Yan
Journal of Organometallic Chemistry 2013 747() pp: 90-97
Publication Date(Web):
DOI:10.1016/j.jorganchem.2013.03.026
Co-reporter:Qibai Jiang;Dan Duan-Mu;Dr. Wei Zhong;Dr. Hao Chen;Dr. Hong Yan
Chemistry - A European Journal 2013 Volume 19( Issue 6) pp:1903-1907
Publication Date(Web):
DOI:10.1002/chem.201203856
Co-reporter:Xiaolei Zhang, Xiao Tang, Jiajia Yang, Yizhi Li, Hong Yan, and Vladimir I. Bregadze
Organometallics 2013 Volume 32(Issue 6) pp:2014-2018
Publication Date(Web):March 1, 2013
DOI:10.1021/om400097m
A new o-carborane-based boron-substituted trithiol has been prepared via direct attachment of sulfur atoms to the B8/B9/B12 vertexes of o-carborane. On the basis of the trithiolate ligand, both the neutral 51 cluster valence electron (CVE) and the cationic 50-CVE trimeric cobalt clusters have been synthesized and structurally characterized. Electrochemical observations demonstrate that the 51-CVE cluster is electron-overloaded and readily undergoes one-electron oxidation to yield the thermodynamically stable 50-CVE species.
Co-reporter:Chao Shi;Huibin Sun;Xiao Tang;Wen Lv;Dr. Hong Yan;Qiang Zhao;Jingxia Wang;Wei Huang
Angewandte Chemie International Edition 2013 Volume 52( Issue 50) pp:13434-13438
Publication Date(Web):
DOI:10.1002/anie.201307333
Co-reporter:Chao Shi;Huibin Sun;Xiao Tang;Wen Lv;Dr. Hong Yan;Qiang Zhao;Jingxia Wang;Wei Huang
Angewandte Chemie 2013 Volume 125( Issue 50) pp:13676-13680
Publication Date(Web):
DOI:10.1002/ange.201307333
Co-reporter:Rui Zhang ; Lin Zhu ; Guifeng Liu ; Huimin Dai ; Zhenzhong Lu ; Jianbo Zhao
Journal of the American Chemical Society 2012 Volume 134(Issue 25) pp:10341-10344
Publication Date(Web):June 15, 2012
DOI:10.1021/ja303334t
We report the one-pot reactions of the 16e– half-sandwich complex CpCoS2C2B10H10 (1), methyl propiolate, and 3e–-donor ligands, which lead to selective B-functionalization at carborane with cyclopentadienyl as a functional group at ambient temperature in good yields. Metal-promoted activations of both a B–H bond of the carborane and a C–H bond of the Cp unit take place sequentially in the cooperation of organic ligands. The reaction requires a 3e–-donor ligand and an activated alkyne and is therefore suitable for a broad range of substrates. This investigation provides a simple and efficient synthetic route to B-functionalized carborane derivatives.
Co-reporter:Rui Zhang, Lin Zhu, Zhenzhong Lu, Hong Yan and Vladimir I. Bregadze
Dalton Transactions 2012 vol. 41(Issue 39) pp:12054-12063
Publication Date(Web):03 Aug 2012
DOI:10.1039/C2DT31425H
The reaction of the 16-electron half-sandwich complex MeCpCo(S2C2B10H10) (1b; MeCp = methylcyclopentadienyl) and methyl propiolate (HCCCO2Me) at ambient temperature leads to MeCpCo(S2C2B10H9)(CHCHCO2Me) (2), MeCpCo(S2C2B10H8)(CHCHCO2Me)2 (3), MeCpCo(S2C2B10H9)[MeO2CCCH(MeO2C)CCH)](CHCHCO2Me) (4) and MeCpCo(S2C2B10H9)(CH2CCO2Me) (5). The reaction of Me4CpCo(S2C2B10H10) (1c; Me4Cp = tetramethylcyclopentadienyl) and the alkyne gives rise to Me4CpCo(S2C2B10H10)[MeO2CCCH(MeO2C)CCH] (6) and Me4CpCo (S2C2B10H9)(CH2CCO2Me) (7). Both 2 and 3 are 16-electron complexes but containing a B(3)-substituted o-carborane-1,2-dithiolate ligand in 2 and a B(3,6)-disubstituted o-carborane-1,2-dithiolate ligand in 3, respectively. In 4 and 6, two alkynes are inserted into one Co–S bond to meet an 18 electron configuration at metal, however, 4 has one B-substitution at carborane. Both 5 and 7 have the same structural type bearing a B–CH2 unit. The reactions of Cp#Co(E2C2B10H10) [Cp# = Cp (1a), MeCp (1b), Me4Cp (1c) and Me5Cp (1d); E = S, Se] with 2-methylpropanedithioic acid (L1) or pyrrolidine-1-carbodithioic acid (L2) lead to Co[S2CCH(CH3)2]3 (8) or Co[S2CN(CH2)4]3 (9), respectively, in an octahedral geometry. The three-component reactions of 1a–1d, methyl propiolate and L1 or L2 afford seven new compounds Cp#Co[S2C2B10H10(CHCHCO2Me)][S2CCH(CH3)2] [Cp# = Cp (10a), MeCp (10b) and Me4Cp (10c)], [S2CCH(CH3)2]2Co(S2C2B10H10)(CHCCO2Me){CpCo[S2CCH(CH3)2]} (11a), Cp#Co[S2C2B10H10(CHCHCO2Me)][S2CN(CH2)4] [Cp# = Cp (12a), MeCp (12b) and Me4Cp (12c)]. All 10a–10c and 12a–12c contain one deprotonated L1 or L2 ligand and one reduced alkyne. 11a has two 18-electron Co centers linked by one reduced alkyne. One metal is coordinated by an o-carborane-1,2-dithiolate and two L1 ligands, and the other is coordinated by one L1 ligand and one η5-Cp unit. In both two- and three-component reactions the reactivity of the 16-electron half-sandwich complexes Cp#Co(S2C2B10H10) is dependent on the size of the Cp# unit. All compounds were fully characterized by spectroscopic techniques and elemental analysis. The solid-state structures of 4, 5, 7, 10a and 11a were further determined by X-ray crystallographic analysis.
Co-reporter:Huan Dou, Wei Zhong, Liu Yang, Tingting Wang, Hong Yan, Yayi Hou
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 15) pp:4693-4700
Publication Date(Web):1 August 2012
DOI:10.1016/j.bmc.2012.06.016
Two half-sandwich cobalt and rhodium complexes 2a and 2b with combination of carborane and N-Sulfonamide were synthesized and fully characterized by NMR spectroscopy, mass spectrometry, elemental analysis as well as X-ray crystallography. In an in vitro cytotoxicity assay toward the non-small cell lung cancer cell lines of A549 and NCI-H460, 2b showed the stronger activity than 2a, which was confirmed by the morphological test. Mechanistic studies for 2b showed that inhibition of NSCLC cell growth was mediated by G0/G1 cell cycle arrested without the significant apoptosis induction. Furthermore, 2b altered the mRNA levels of CCND1, CCNE1 and PCNA, which were known to control G0/G1 phase of the cell cycle. Our western blot analysis also showed that 2b-induced G0/G1 cell cycle arrest was mediated through the decreased expression of cyclin D1, cyclin E1 and PCNA.
Co-reporter:Jiurong Hu, Xiao Tang, Guifeng Liu, Qibai Jiang, Dandan Duanmu, Yizhi Li, Hong Yan
Journal of Organometallic Chemistry 2012 s 721–722() pp: 36-41
Publication Date(Web):
DOI:10.1016/j.jorganchem.2012.04.015
Co-reporter:ChunHui Wu;HongDe Ye;Hui Jiang;XueMei Wang
Science China Chemistry 2012 Volume 55( Issue 4) pp:594-603
Publication Date(Web):2012 April
DOI:10.1007/s11426-011-4490-9
The interactions between the new organometallic complexes, ferrocene-substituted dithio-o-carborane conjugates (denoted as FcSB1, FcSB2 and FcSBCO) and hemoglobin (Hb) are investigated by electrochemistry, fluorescence and UV-vis absorption spectroscopy. The results demonstrate that FcSB1, FcSB2 and FcSBCO can bind to the heme iron center through the replacement of the weakly bound H2O/O2 in the distal heme pocket of Hb by their sulfur donor atoms, inducing the allosteric change from the R state (oxygenated conformation, relax) to T state (deoxygenated conformation, tense). The binding affinity is in the order of FcSBCO>FcSB2>FcSB1. Moreover, the fluorescence study illustrates that the three ferrocene-carborane conjugates differently affect the quarterly and tertiary structures as well as the polarity in the surrounding of the Trp and Tyr residues in Hb. Typically, FcSB2 mainly induces alterations of the microenvironment around the β37Trp residue which is located on the α1β2 interface of Hb. Such distinct influences are attributed to the structural features of FcSB1, FcSB2 and FcSBCO containing hydrophobic ferrocenyl and carboranyl units as well as C=O group. Screening the protein-binding behavior can signify the potential bioactivity of such molecules and may be helpful in the future development of promising multifunctional metallodrugs.
Co-reporter:Wei Zhong, Qiwu Yang, Yi Shang, Guifeng Liu, Haitao Zhao, Yizhi Li, and Hong Yan
Organometallics 2012 Volume 31(Issue 18) pp:6658-6668
Publication Date(Web):September 6, 2012
DOI:10.1021/om300735d
The reactions of the 16-electron half-sandwich complex CpCo(S2C2B10H10) (1) (Cp: cyclopentadienyl) with sulfonyl azides (p-toluenesulfonyl azide, TsN3; methanesulfonyl azide, MsN3) in refluxing dichloromethane or at ambient temperature lead to imido-bridged adducts CpCo(S2C2B10H10) (NSO2R) (2a, R = 4-MePh; 2b, R = Me) which can convert to the tetraazadiene cobalt complexes CpCoN4(SO2R)2 (3a, R = 4-MePh; 3b, R = Me) in the presence of excess azide if heated. The reactions of 1 with acyl azides (methyl azidoformate and benzoyl azide) lead to CpCo(S2C2B10H10)(CONR) (4a, R = OMe; 4b, R = Ph) with a newly-generated five-membered metallacyclic ring Co–S–N–C–O. Complexes 2a and 2b show further reactivity toward alkynes to give rise to the insertion products CpCo(S2C2B10H10)(R1C═CR2) (NSO2R) (R1 = COOMe, R2 = H, R = 4-MePh, 5a, R = Me, 5b; R1 = R2 = COOMe, R = 4-MePh, 6a, R = Me, 6b; R1 = COOMe, R2 = Ph, R = 4-MePh, 8a, R = Me, 8b) formed by alkyne addition to a Co–S bond to generate a Co–C–C–S four-membered ring and CpCo(S2C2B10H10)(R1C═CR2NSO2R) (R1 = H, R2 = Ph, R = 4-MePh, 7a, R = Me, 7b; R1 = COOMe, R2 = Ph, R = 4-MePh, 9a, R = Me, 9b) formed by alkyne insertion into a Co–N bond to generate a Co–C–C–N–S five-membered ring. In the case of PhC≡CCO2Me, the products with insertion into both Co–S and Co–N bonds are isolated. Interestingly, if tert-butylacetylene is used, CpCo(S2C2B10H10)(R1R2C═CNSO2R) (R1 = tBu, R2 = H, R = 4-MePh, 10a, R = Me, 10b) are generated by insertion of terminal carbon into a Co–N bond to form four-membered ring Co–C–N–S. The insertion pathways of these reactions have been discussed on the basis of DFT calculations. All the new complexes were fully characterized, and X-ray structural analyses were performed for 2a, 3a, 3b, 4a, 4b, 5a, 6a, 7a, 7b, 8a, 9a, 9b, and 10b.
Co-reporter:Guifeng Liu, Jiurong Hu, Jialin Wen, Huimin Dai, Yizhi Li, and Hong Yan
Inorganic Chemistry 2011 Volume 50(Issue 9) pp:4187-4194
Publication Date(Web):March 28, 2011
DOI:10.1021/ic200333q
The reaction of the 16-electron half-sandwich complex CpCo(S2C2B10H10) (1; Cp = cyclopentadienyl) with ethyl diazoacetate (EDA) at ambient temperature leads to compounds CpCo(S2C2B10H10)(CHCO2Et) (2), CpCo(S2C2B10H8)(CHCO2Et)(CH2CO2Et)[CH(CO2Et)(CH2CO2Et)] (3), CpCo(S2C2B10H9)(CH2CO2Et)(CHCO2Et)2 (4), CpCo(S2C2B10H9)(CHCO2Et)(CH2CO2Et) (5), and CpCo(S2C2B10H9)(CHCO2Et)2(CH2CO2Et) (6). In 2, the EDA molecule has been inserted into one Co−S bond in 1 with the loss of N2 to form an 18-electron compound containing a three-membered metallacyclic ring. In 3, two B−H bonds of the carborane cage have been activated and the unusual B4−H bond activation leads to the formation of a stable Co−B bond. Two EDA molecules are inserted into the Co−B3 bond to generate an unexpected six-membered heterocyclic ring Co−B−B−C−C−O. In 4, a stable Co−B bond is present as well but in the position B3/B6, and two EDA molecules are inserted into one Co−S bond to produce a five-membered heterocyclic ring Co−C−C−C−O. In 5, one EDA is inserted into the Co−B bond with the formation of a C−B bond in the position B3/B6. One more EDA is inserted into the Co−S bond in 5 to generate 6. Upon heating, 6 loses the BH vertex close to the two carbon atoms to lead to CpCo(S2C2B9H9)(CHCO2Et)(CH2CO2Et)2 (7) containing a nido-C2B9 unit. All of the new compounds 2−7 were characterized by NMR spectroscopy (1H, 11B, and 13C), mass spectrometry, IR spectroscopy, and elemental analysis, and their solid-state structures were further characterized by X-ray structural analysis.
Co-reporter:Chunhui Wu, Hongde Ye, Wenjuan Bai, Qingning Li, Dadong Guo, Gang Lv, Hong Yan, and Xuemei Wang
Bioconjugate Chemistry 2011 Volume 22(Issue 1) pp:16
Publication Date(Web):December 16, 2010
DOI:10.1021/bc100158b
The large diversity of structures and unique bonding modes of organometallic complexes make them possible to act as promising candidate therapeutic agents. In this study, the new type of ferrocene-substituted dithio-o-carborane conjugates (FcSB1, FcSB2, and FcSBCO) has been synthesized, and their in vitro antineoplastic activities have been explored by means of the electrochemical study, the real time cell electronic sensing (RT-CES) system, and biological assays. The conjugate−cell interactions were first monitored by electrochemistry, and the results show different cell uptake efficiency for FcSB1, FcSB2, and FcSBCO toward target cells. Both the highly hydrophobic ferrocenyl and carboranyl groups render the conjugates able to rapidly enter cells and exert acute cytotoxicity after 4 h incubation in serum-free media. However, FcSB1, FcSB2, and FcSBCO display different inhibition efficiencies toward SMMC-7721 and HepG2 cancer cells via the G0/G1 arrest mechanism in a physiological environment. The anticancer activity is in the order FcSB2 > FcSB1 > FcSBCO, which is parallel to the order of the redox potentials of the ferrocenyl groups in the three complexes. In particular, FcSB1 and FcSB2 display a potent selective inhibition effect on the proliferation of the cancer cell lines SMMC-7721 and HepG2, but almost no effect on the normal cell line, the human embryonic lung fibroblast (HELF) cells. Thus, these results may provide some clues for use of the ferrocene−carborane conjugates in developing anticancer drugs.
Co-reporter:Zhi-Wei Xu, Lei Han, Cheng Ji, Rui Zhang, Xu-Jie Shen and Hong Yan
Dalton Transactions 2011 vol. 40(Issue 26) pp:6992-6997
Publication Date(Web):06 Jun 2011
DOI:10.1039/C1DT10291E
The reactions of the 16e half-sandwich complex (p-cymene)Ru(S2C2B10H10) (Ru16e) with 1,4-diethynylbenzene (L1), 3′,6-diethynyl-1,1′-binaphthyl-2,7′-diyl diacetate (L2), 2-bromo-5-ethynylthiophene (L3) and 2,5-diethynylthiophene (L4) lead to 18e mononuclear complexes (p-cymene)Ru(S2C2B10H9)(H2CCPhCCH) (1), (p-cymene)Ru(S2C2B10H9)[H2CC(C24H16O4)CCH] (2), (p-cymene)Ru(S2C2B10H9) [H2CC(C4H2S)Br] (3) and (p-cymene)Ru(S2C2B10H9) [H2CC(C4H2S)CCH] (4), respectively. In all of them, metal-induced B–H activation has occurred, which leads to a stable Ru–B bond, and the structures take a cisoid arrangement. Only in the case of L4, the binuclear complexes [(p-cymene)Ru(S2C2B10H9)]2[H2CC(C4H2S)CCH2] (5a and 5b) are observed, which are conformational isomers generated by the differing orientations of the p-cymene unit. 4 can be readily converted to the complex (p-cymene)Ru(S2C2B10H9)[H2CC(C4H2S)COCH3] (6) in the presence of silica and H2O. All of these products 1–6 were characterized by NMR, IR, elemental analysis and mass spectrometry. The structures of 1, 3, and 5a were also determined by single-crystal X-ray diffraction analysis.
Co-reporter:Hongde Ye, Baohua Xu, Mingshi Xie, Yizhi Li and Hong Yan
Dalton Transactions 2011 vol. 40(Issue 24) pp:6541-6546
Publication Date(Web):17 May 2011
DOI:10.1039/C1DT10315F
The reaction of the dinuclear cobalt compound [(CpCoS2C2B10H10)(CpCoSC2B10H11)(n-C4H9S)] (1) with HCCC(O)Fc leads to the cobalt-free products (C2B10H10)(SCHCHCOFc)2 (4–6), (S2C2B10H10)(HCCCOFc) (7), and (C2B10H11)(SCHCHCOFc) (8, 9). 4–6 are produced by hydrosulfuration of the alkyne at the 1,2-dicarba-closo-dodecaborane-dithiolate ligand with the generated vinyl groups in Z/Z, Z/E and E/E configurations, respectively. In 7, the alkyne is added to 1,2-dicarba-closo-dodecaborane-dithiolate at the two sulfur sites. 8 and 9 are the products of alkyne hydrosulfuration at the 1,2-dicarba-closo-dodecaborane-1-monothiolate ligand with the generated vinyl group in either Z or E configuration. The treatment of 1 with HCCCO2Me gives rise to the parallel products (C2B10H10)(SCHCHCO2Me)2 (10–12) and (C2B10H11)(SCHCHCO2Me) (13, 14). All of the new compounds have been characterized by IR, NMR, elemental analysis and mass spectroscopy. The structures of compounds 4, 7, and 8 have also been determined by single-crystal X-ray diffraction analysis.
Co-reporter:Hong-De Ye, Guan-Yu Ding, Ming-Shi Xie, Yi-Zhi Li and Hong Yan
Dalton Transactions 2011 vol. 40(Issue 10) pp:2306-2313
Publication Date(Web):12 Nov 2010
DOI:10.1039/C0DT00933D
The 16e half-sandwich complex Cp*Co(S2C2B10H10)(1, Cp* = pentamethylcyclopentadienyl) reacts with N-1-naphthylpropargylamide to afford two 18e complexes (2) and (3). 2 is a 1:1 adduct containing a four-membered metallacycle. In 3 the alkyne is twofold inserted into one Co–S bond in a head–head mode. Reactions of 1 with 1-(2-furyl)-2-propyn-1-one and 1-ferrocenyl-2-propyn-1-one lead to two types of alkyne twofold inserted products (4)/(5) and (6)/(7), respectively. The reaction of 1 with phenylacetylene gives rise to the sole complex (8) of the same structural type as 3, 4 and 6, whereas the reaction of 1 with dimethyl acetylene dicarboxylate affords the sole 1:1 adduct (9). Complexes 2–9 have been characterized by IR, NMR, elemental analysis and mass spectrometry, and 3–9 have also been determined by single-crystal X-ray diffraction analysis.
Co-reporter:Hongde Ye;Wenjun Bai;Mingshi Xie;Yizhi Li
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 17) pp:2763-2768
Publication Date(Web):
DOI:10.1002/ejic.201100159
Abstract
The 16-electron half-sandwich complex CpCo(Se2C2B10H10) (1, Cp = cyclopentadienyl) reacts with FcC(O)C≡CH in CH2Cl2 at ambient temperature to give [{FcC(O)CCH}(Se2C2B10H10)] (3) and [CpCo(Se2C2B10H9){CH2CC(O)Fc}] (4). Complex 3 is generated by the alkyne addition to the 1,2-dicarba-closo-dodecaborane-1,2-diselenolate ligand in 1 with the loss of the CpCo unit. In 4, metal-induced B–H activation occurs in the B(3)/B(6) position of carborane followed by the formation of a C–B bond. In MeOH the reaction leads to an unexpected product, [{FcC(O)CHCH}CpCo(Se2C2B9H9){CH2CC(O)Fc}] (5), which contains two alkynes and a nido-C2B9 cage. In MeOH, 4 can further react with the alkyne to give 5. In the presence of silica, 4 loses the CpCo unit to afford [{FcC(O)CCH2}(Se2C2B10H9)] (6), which is a symmetrical molecule with the B–CH2 unit retained. All of these complexes have been characterized by IR, NMR, elemental analysis, mass spectrum, and single-crystal X-ray diffraction analysis.
Co-reporter:Jiurong Hu, Jialin Wen, Dehong Wu, Rui Zhang, Guifeng Liu, Qibai Jiang, Yizhi Li, and Hong Yan
Organometallics 2011 Volume 30(Issue 2) pp:298-304
Publication Date(Web):December 8, 2010
DOI:10.1021/om1010275
Treatment of o-carborane, n-butyllithium, selenium, and [(p-cymene)RuCl2]2 under argon leads to the complexes (p-cymene)Ru(Se2C2B10H10) (1), [(p-cymene)RuCl(Se−SeC2B10H11)][(p-cymene)Ru(Se2C2B10H10)] (2), [(p-cymene)Ru2(Se2C2B10H10)2][(p-cymene)Ru(Se2C2B10H9)] (3), and [(p-cymene)Ru(Se2C2B10H9) Ru(Se2C2B10H10)]2 (4). 2 contains a rhombic Ru2Se2 core and can be considered as an adduct of the two 16e half-sandwich monomers (p-cymene)Ru(Se2C2B10H10) (1) and (p-cymene)RuCl(Se−SeC2B10H11). In 3 three o-carborane-1,2-diselenolate ligands bridge the adjacent Ru atoms and one [Se2C2B10H9]3− unit is in a tridentate binding mode with a Ru−B bond. In 4, two identical dinuclear (p-cymene)Ru2(Se2C2B10H9)(Se2C2B10H10) subunits connect to each other through two Se atoms of two individual [Se2C2B10H10]2− ligands and a metal-induced B−H activation occurs as well. However, if the above reaction mixture is exposed to air, the unexpected product [(p-cymene)Ru]2(Se2C2B9H10)Ru2(Se2C2B10H10)3(μ-O) (5) can be isolated in moderate yield. It contains a novel Ru4Se8O core generated from three [Se2C2B10H10]2−, one nido-[Se2C2B9H10]3−, and one μ-oxo unit. Complexes 2−5 have been characterized by elemental analyses, mass and NMR spectra, and X-ray crystallography.
Co-reporter:Wenjuan Bai, Guifeng Liu, Gulnisa Guoyiqibayi, and Hong Yan
Organometallics 2011 Volume 30(Issue 19) pp:5188-5195
Publication Date(Web):September 16, 2011
DOI:10.1021/om2005518
The reaction of the 16-electron (16e) half-sandwich complex CpCo(S2C2B10H10) (1; Cp = cyclopentadienyl) with ethynylferrocene (Fc–C≡CH) in CH2Cl2 at ambient temperature leads to (CHCFc)(S2C2B10H10) (2), where the alkyne is added to the o-carborane-1,2-dithiolate ligand at the two sulfur sites with the loss of the CpCo unit. However, in MeOH the reaction gives rise to an additional product, CpCo(S2C2B9H9)(CH2CFc)(CH2═CFc) (3), in which one alkyne has been reduced to an alkane containing a B–CH2 unit and the second alkyne is present as a terminal alkene at the second sulfur site. If silica and moisture are present, CpCo(S2C2B9H10)(CH═CFc)(CH2C(O)Fc) (4) could be generated, in which one alkyne is stereoselectively inserted into one Co–S bond and the second alkyne is present as an acetyl ferrocenyl group at the second sulfur site. Both 3 and 4 contain a nido-C2B9 unit. Moreover, in boiling toluene the reaction leads to CpCo(S2C2B10H10)(CHCFc) (5), where the cleavage of the weaker Co–C and C–S bonds and the formation of a stronger C–C bond has occurred. In comparison, the reaction of Cp*Co(S2C2B10H10) (1*; Cp* = pentamethylcyclopentadienyl) at ambient temperature gives rise to the alkyne 2-fold inserted product Cp*Co(S2C2B10H10)(FcCCHCHCFc) (6). If 6 is heated in boiling toluene, the unusual product (FcCSCHCHCFc)(SC2B10H10) (7) is obtained, where two alkynes are dimerized with the formation of two five-membered S-heterocycles and the loss of a Cp*Co unit. The five new compounds 3–7 were characterized by NMR (1H, 11B, 13C), mass spectroscopy, and X-ray structural analysis.
Co-reporter:Yuguang Li ; Qibai Jiang ; Yizhi Li ; Hong Yan ;Vladimir I. Bregadze
Inorganic Chemistry 2010 Volume 49(Issue 1) pp:4-6
Publication Date(Web):December 4, 2009
DOI:10.1021/ic901970b
The reaction of the 16e half-sandwich complex CpCo(S2C2B10H10) (1) with HC≡C−C(O)Ph leads to a 17e complex 2 containing a bicyclo[2.2.1]heptene unit at the B3/B6 site of carborane. In the presence of air, water and silica 2 converts to two 16e complexes, 3 and 4, bearing new types of functionalized groups.
Co-reporter:Yuguang Li ; Qibai Jiang ; Xiaolei Zhang ; Yizhi Li ; Hong Yan ;Vladimir I. Bregadze
Inorganic Chemistry 2010 Volume 49(Issue 8) pp:3911-3917
Publication Date(Web):March 19, 2010
DOI:10.1021/ic1001804
The reactions of the 16e half-sandwich complex, CpCo(S2C2B10H10) (1), with alkynones at ambient temperature lead to complexes CpCo(S2C2B10H9)(CH═CH−C(O)R) (R = Ph (2), 2-naphthyl (3)). Both 2 and 3 are still 16e half-sandwich complexes containing a B(3)-substituted ortho-carborane-1,2-dithiolate ligand. Treatment of 2 with excess alkynes R1C≡CR2 (R1 = H, R2 = Ph, C(O)Ph, CO2Me; R1 = R2 = CO2Me) affords five complexes, CpCo(S2C2B10H8)(CH2CPh)(CH═CH−C(O)Ph) (4), CpCo(S2C2B10H8)(CH═CH−C(O)Ph)2 (5), CpCo(S2C2B10H8)(CH═CH−CO2Me)(CH═CH−C(O)Ph) (6), CpCo(S2C2B10H9)(MeO2C−C═C−CO2Me)(CH═CH−C(O)Ph) (7), and CpCo(S2C2B10H9)(MeO2C−C═C−CO2Me)2(CH═CH−C(O)Ph) (8). Complex 4 is an 18e complex bearing a B−CH2 unit. Both 5 and 6 retain a 16e half-sandwich structure but contain a B(3,6)-disubstituted ortho-carborane-1,2-dithiolate ligand. However, in 7 and 8 one or two alkynes are inserted into one of the Co−S bonds to generate 18e species, respectively. Heating 7 leads to the 16e complex, CpCo(S2C2B10H8)(MeO2C−C═CH−CO2Me)(CH═CH−C(O)Ph) (9), having a B(3,6)-disubstituted ortho-carborane-1,2-dithiolate ligand as in 5 and 6. All complexes were fully characterized by spectroscopic techniques and elemental analysis. The solid-state structures of 2 and 3 and 5−9 were further characterized by X-ray structural analysis.
Co-reporter:Yuguang Li ; Qibai Jiang ; Yizhi Li ; Xujie Shen ; Hong Yan ;Vladimir I. Bregadze
Inorganic Chemistry 2010 Volume 49(Issue 12) pp:5584-5590
Publication Date(Web):May 4, 2010
DOI:10.1021/ic100497h
Treatment of ortho-carborane, n-butyl lithium, sulfur, and CpCo(CO)I2 with differing ratios led to four new compounds [(CpCoS2C2B10H10)(CpCoSC2B10H11)(SC4H9)] (3), [(CpCo)2S2(S2C2B10H10) (SC2B10H10)(SC4H9)] (4), [(CpCo)2Co(SC4H9)6]+[Co(S2C2B10H10)2]− (5), and [(CpCo)2Co(SC2B10H11)(SC4H9)5]+[Co(S2C2B10H10)2]− (6). Compound 3 contains a rhombic Co2(μ2-S)2 core and can be considered as an adduct of the two 16e half-sandwich monomers CpCo(S2C2B10H10) (1) and CpCo(SC4H9)(SC2B10H11). Compound 4 is a trinuclear complex containing a cuboidal [Co3(μ3-S)2(μ-S)2] core, and one cobalt atom is coordinated by six sulfur atoms in a distorted octahedral geometry. Compound 5 consists of a linear-type trinuclear cobalt(III) monocation bridged by six n-butyl thiolato units and a square-planar cobalt(III) monoanion [Co(S2C2B10H10)2]−. However, in 6, one ortho-carborane monothiolato ligand [SC2B10H11]− has replaced one n-butyl thiolato ligand in 5. This leads to a nonlinear arrangement of the three cobalt atoms in the cation and the nonplanar [Co(S2C2B10H10)2]− anion in 6. Interestingly, the treatment of the 16e half-sandwich CpCo(S2C2B10H10) (1) by [N(n-Bu)4]Br afforded the ionic compound 7, [N(n-Bu)4]+[Co(S2C2B10H10)2]−, containing a square-planar anion.
Co-reporter:Jiurong Hu, Guifeng Liu, Qibai Jiang, Rui Zhang, Wei Huang, and Hong Yan
Inorganic Chemistry 2010 Volume 49(Issue 23) pp:11199-11204
Publication Date(Web):October 28, 2010
DOI:10.1021/ic101817u
Treatment of ortho-carborane, n-butyl lithium, sulfur, and [(p-cymene)RuCl2]2 in varying ratio led to four new compounds (p-cymene)Ru[S3(C2B10H10)2] (3), [(p-cymene)Ru2(μ2-S2C2B10H9)(μ3-S2C2B10H10)]2 (4), [(p-cymene)Ru]2Ru(μ2-η2:η2-S2) (μ2-η2:η1-S2Cl)(μ2-S2C2B10H10)2 (5), and [(p-cymene)Ru]2Ru(μ2-η1:η1-S2)(μ3-η2:η2-S4) (μ2-S2C2B10H10)2 (6), respectively. In 3, the ruthenium atom is coordinated by three S atoms from a in situ generated tridentate [S3(C2B10H10)2]2− ligand. 4 consists of two identical dinuclear (p-cymene)Ru2(μ2-S2C2B10H9)(μ3-S2C2B10H10) subunits which connect to each other via the Ru−Ru bond and two bridging o-carborane-1,2-dithiolate ligands. In 4, a Ru−B bond is present. 5 contains a Ru3(μ2-S)2(μ2-S2)(μ2-S2Cl) core, and the central ruthenium atom is coordinated by seven S atoms in a distorted pentagonal bipyramidal geometry. In 5, a S−Cl bond is generated. 6 has a novel Ru3(μ2-S)2(μ2-S2)(μ3-S4) core, and the three ruthenium atoms are connected through the two terminal sulfur atoms of the S−S−S−S chain in a μ3 binding fashion. All the four complexes have been characterized by elemental analysis, mass, NMR, and X-ray crystallography.
Co-reporter:Chunhui Wu;Baohua Xu Dr.;Juan Zhao;Qibai Jiang;Fadong Wei;Hui Jiang Dr.;Xuemei Wang
Chemistry - A European Journal 2010 Volume 16( Issue 29) pp:8914-8922
Publication Date(Web):
DOI:10.1002/chem.201000605
Abstract
Biointeractions between two organometallic compounds, a pair of ferrocene-substituted dithio-o-carborane isomers (C14H20B10FeS2; denoted as FcSB1 and FcSB2), and myoglobin (Mb) have been investigated by means of electrochemistry, fluorescence, circular dichroism, and UV/Vis absorption spectroscopy. Our observations demonstrate that FcSB1 and FcSB2 could coordinate to the axial position trans to the histidine imidazole that induces the change of the heme iron from the high spin state to the low spin state and the changes of the conformation of the aromatic fluorophores of the selected heme protein. Such influences attribute to the structural features of FcSB1 and FcSB2 containing sulfur donor atoms and hydrophobic ferrocenyl and carboranyl units that leads to specific binding modalities with Mb. This study provides an insight into the understanding of relevant biointeractions between the new type of ferrocene–carborane conjugates and hemoproteins, and might shed light on the promising bioapplications of these multifunctional organometallic complexes.
Co-reporter:YuGuang Li;HongDe Ye;Gulnisa Guoyiqibayi;QiBai Jiang
Science China Chemistry 2010 Volume 53( Issue 10) pp:2129-2138
Publication Date(Web):2010 October
DOI:10.1007/s11426-010-4103-z
The reaction of the 16e half-sandwich complex Cp*Co(S2C2B10H10) (1) (Cp* = pentamethylcyclopentadienyl) with excess methyl acetylene monocarboxylate, HC≡C-CO2Me, affords the 18e complexes 2–6. Compound 2 bears a B-CH2 unit in which B-substitution occurs in the B(3)/B(6) position of the ortho-carborane cage. Complexes 3–6 are geometrical isomers, in which the alkyne is twofold inserted into one of the Co-S bonds in all the four possible ways. Treatment of 1 with excess 3-butyn-2-one or phenyl ethynyl ketone, HC≡C-C(O)R (R = Me, Ph), at ambient temperature leads to the 18e complexes 7–10, respectively, with two alkynes inserted into one of the Co-S bonds. All the new complexes were fully characterized by spectroscopic techniques and elemental analysis. The solid-state structures of 2, 3, 5, 7, 8, 9 and 10 were further characterized by X-ray structural analysis.
Co-reporter:Chun-Hui Wu, De-Hong Wu, Xuan Liu, Gulnisa Guoyiqibayi, Da-Dong Guo, Gang Lv, Xue-Mei Wang, Hong Yan, Hui Jiang and Zu-Hong Lu
Inorganic Chemistry 2009 Volume 48(Issue 6) pp:2352-2354
Publication Date(Web):February 16, 2009
DOI:10.1021/ic900009j
Two new ruthenium(II) arene complexes, 2a (C24H34B10FeRuS2) and 2b (C15H26B10O2RuS2), bearing a carborane unit and other different functional groups were synthesized, and their cytostatic effects on cancerous cells were evaluated. Our observations illustrate that a structural change from a ferrocene unit to a carboxyl group could lead to high selectivity toward cancer cells and facilitate the efficient inhibition of the proliferation of target cells, indicating that the tuning of the overall properties of the ruthenium(II) arene complex by appropriate ligand tagging is critical to creating a selective antineoplastic agent.
Co-reporter:De-Hong Wu, Chun-Hui Wu, Yi-Zhi Li, Da-Dong Guo, Xue-Mei Wang and Hong Yan
Dalton Transactions 2009 (Issue 2) pp:285-290
Publication Date(Web):10 Nov 2008
DOI:10.1039/B810831E
The addition reactions of the 16e half-sandwich complexes (p-cymene)M(S2C2B10H10) (1S, M = Ru; 2S, M = Os) and Cp*Ir(E2C2B10H10) (3S, E = S; 3Se, E = Se) with ethynylferrocene lead selectively to the 18e complexes (p-cymene)Ru(S2C2B10H9)(H2CCFc) (Fc = ferrocenyl) (4S), (p-cymene)Os(S2C2B10H9)(H2CCFc) (5S), Cp*Ir(S2C2B10H9)(H2CCFc) (6S) and Cp*Ir(Se2C2B10H9)(H2CCFc) (6Se), in which the alkyne is regio- and stereoselectively inserted into one of the M–E bonds that may further lead to metal-induced B–H activation, hydrogen atom transfer from the carboranevia the metal center to the inserted alkyne, and the generation of a M–B bond. In all complexes the S-η2-(Fc)C–C and C–B(M) moieties occupy a cisoid position. The four new complexes are characterized by IR, MS, NMR spectroscopy and microanalysis, and the X-ray structural analysis of 4S is performed. 4S was observed to promote the uptake of anticancer drug daunorubicin in drug-resistant leukemia K562 cells.
Co-reporter:Bao-Hua Xu ; Xu-Qing Peng ; Zhi-Wei Xu ; Yi-Zhi Li
Inorganic Chemistry 2008 Volume 47(Issue 17) pp:7928-7933
Publication Date(Web):August 5, 2008
DOI:10.1021/ic801199e
Disulfuration and hydrosulfuration of alkynes mediated by an unusual square-planar tetrathiolate cobalt(III) complex [Cp2Co]+[Co(S2C2B10H10)2]−, 1, lead to a series of cobalt-free carboranyl vinyl sulfides 2−9. All new complexes 1, 2, 3, 4, 5, 6, 7, 8, and 9 were characterized by NMR spectroscopy (1H, 11B, 13C), and X-ray structural analyses were performed for 1, 2, 3, 5, 6, 7, and 9.
Co-reporter:Dehong Wu ; ; Yuguang Li ; ; Lei Han ; ; Yizhi Li ; ;
Inorganic Chemistry 2008 Volume 47(Issue 14) pp:6524-6531
Publication Date(Web):June 3, 2008
DOI:10.1021/ic800619z
Treatment of 1a and 1b with phenylacetylene, 1,4-diethynylbenzene, and ethynylferrocene affords addition complexes, ( p-cymene)Ru(μ-E 2)Ru(S 2C 2B 10H 10) 2(R 1C=CR 2), (R 1 = Ph (H), R 2 = H (Ph), 2a, 2b, 3a, 3b; R 1 = −Ph−CCH (H), R 2 = H (−Ph−CCH), 4a, 4b, 5a, 5b; R 1 = Fc (H), R 2 = H (Fc), 6a, 6b, 7a, 7b). Alkyne addition occurs at sulfur atoms of two different chelating 1,2-dicarba- closo-dodecaborane-1,2-dithiolate ligands that leads to a change of 16e Ru(IV) in 1a and 1b to 18e Ru(II) in 2a− 7a and 2b− 7b, respectively. Moreover, the reaction of 1a with ethynylferrocene produces an unprecedented tetranuclear mixed-valent Ru(IV)Ru(II) 3S 12 cluster ( 8a) consisting of two 1a and one ethynylferrocene through the cleavage and generation of Ru−S, S−S, and C−S bonds. The complexes were characterized by IR, MS, and NMR spectroscopy and microanalysis. X-ray structural analyses were performed on 1b, 2a, 3b, 5a, 5b, 7b, and 8a.
Co-reporter:Bao-Hua Xu Dr.;Xu-Qing Peng;Yi-Zhi Li
Chemistry - A European Journal 2008 Volume 14( Issue 30) pp:9347-9356
Publication Date(Web):
DOI:10.1002/chem.200801136
Abstract
The reaction of the 16e half-sandwich complex [CpCo(S2C2B10H10)] (1 S; Cp: cyclopentadienyl) with ethynylferrocene in CH2Cl2 at ambient temperature leads to [CpCo(S2C2B10H9)(CH2CFc)] (2 S; Fc: ferrocenyl) and 1,2,4-triferrocenylbenzene. In 2 S, B substitution occurs at the carborane cage in the position B(3)/B(6) with the formation of a CB bond. In the presence of the protic solvent MeOH, 2 S loses a CpCo fragment to generate [(CH2CFc)(S2C2B10H9)] (3 S). On the other hand, 2 S can take a free CpCo fragment to form [(CpCo)2(S2C2B9H8)(CHCFc)] (4 S) containing a nido-C2B9 unit. In sharp contrast, [CpCo(Se2C2B10H10)] (1 Se) does not react with the alkyne in CH2Cl2, but in MeOH [(CHCFc)(Se2C2B10H10)] (5 Se) is generated without the presence of a CpCo unit. The reaction of 1 with dimethyl acetylenedicarboxylate at ambient temperature leads to insertion compounds [CpCo(E2C2B10H10){(MeO2C)CC(CO2Me)}] (6 S, E=S; 6 Se, E=Se). Upon heating, 6 S rearranges to two geometrical isomers [CpCo(S2C2B10H9){(MeO2C)CCH(CO2Me)}] (7 S) and [CpCo(S2C2B10H9){(MeO2C)CHC(CO2Me)}] (8 S). In both, BH functionalization takes place at the carborane cage in the position B(3)/B(6), but 7 S is a 16e complex with an olefinic unit in a Z configuration, and 8 S is an 18e complex containing an alkyl BCH group. Further treatment of 7 S with dimethyl acetylenedicarboxylate at ambient temperature affords two B-disubstituted complexes at the carborane cage in the positions of the B(3) and B(6) sites, that is, [CpCo(S2C2B10H8){(MeO2C)CCH(CO2Me)}2] (9 S) and [CpCo(S2C2B10H8){(MeO2C)CHC(CO2Me)}{(MeO2C)CCH(CO2Me)}] (10 S). Compound 9 S is a 16e complex with two olefinic units in E/E configurations, whereas 10 S is an 18e species containing both an olefinic substituent and an alkyl BCH unit. The reaction of 7 S with methyl acetylenemonocarboxylate at ambient temperature leads to the sole 16e compound [CpCo(S2C2B10H8){CHCH(CO2Me)}{(MeO2C)CCH(CO2Me)}] (11 S). In contrast, 6 Se does not rearrange. All new complexes 2 S–4 S, 5 Se, 6 Se, and 7 S–11 S were characterized by NMR spectroscopy (1H, 11B, 13C) and X-ray structural analyses were performed for 2 S–4 S, 5 Se, 6 Se, and 7 S–9 S.
Co-reporter:Bao-Hua Xu, Jing-Cong Tao, Yi-Zhi Li, Shu-Hua Li and Hong Yan
Organometallics 2008 Volume 27(Issue 3) pp:334-340
Publication Date(Web):January 11, 2008
DOI:10.1021/om7009864
The reaction of the 16e half-sandwich complex {CpCo[S2C2B10H10]} (1S) with methyl acetylene monocarboxylate at ambient temperature led to {CpCo[S2C2B10H10] [CHC(CO2Me)CHC(CO2Me)]} (2S) and {CpCo[S2C2B10H8][CHCH(CO2Me)]2} (3S). In2S the alkyne is 2-fold inserted into one of the Co−S bonds. 3S is a 16e B-disubstituted complex with the olefinic units in a Z/Z configuration. In comparison, {CpCo[Se2C2B10H10]} (1Se) reacted with the alkyne to give rise to {CpCo[Se2C2B10H10][CHC(CO2Me)CHC(CO2Me)]} (2Se), {CpCo[Se2C2B10H9] [CH2C(CO2Me)]} (4Se), and {CpCo[Se2C2B10H8][CH2C(CO2Me)][CHCH(CO2Me)]} (5Se). 2Se is the analogue of 2S. Upon heating, 2S and 2Se catalyze cyclotrimerization of the alkyne to generate 1,3,5- and 1,2,4-tricarboxylatebenzenes. 4Se is an 18e B-substituted species with a B−CH2 unit. 5Se is analogous to 4Se, but contains an olefinic substituent at the B(3)/B(6) site of the carborane in a Z configuration. Mechanistic implications on metal-induced B−H bond activation and catalytic cyclotrimerization of alkyne were elucidated. All new complexes were characterized by NMR spectroscopy (1H, 11B, 13C), and X-ray structural analyses were reported for 2S, 2Se, 3S, 4Se, and 5Se.
Co-reporter:Xiaolei Zhang, Zhiwen Zhou and Hong Yan
Chemical Communications 2014 - vol. 50(Issue 86) pp:NaN13080-13080
Publication Date(Web):2014/09/05
DOI:10.1039/C4CC06171C
New o-carborane-9,12-dithiolate dirhodium complexes are reported to selectively activate inert B–H bonds of o-carboranes to form B–X bonds by using water, alcohols, and alkylhalides (<60 °C, yields >80%). Characterization of the key intermediates demonstrates an oxidative addition and reductive elimination pathway via metal–metal cooperativity.
Co-reporter:Lin Zhu, Wen Lv, Shujuan Liu, Hong Yan, Qiang Zhao and Wei Huang
Chemical Communications 2013 - vol. 49(Issue 90) pp:NaN10640-10640
Publication Date(Web):2013/09/18
DOI:10.1039/C3CC46276E
Carborane-incorporated (at both carbon and boron sites) tribranched fluorophores were efficiently prepared. oB significantly improves the fluorescence quantum yield. mC exhibits the largest two-photon absorption among the known carborane-based derivatives and has been successfully applied in two-photon fluorescence 2D & 3D bioimaging.
Co-reporter:Rui Zhang, Lin Zhu, Zhenzhong Lu, Hong Yan and Vladimir I. Bregadze
Dalton Transactions 2012 - vol. 41(Issue 39) pp:NaN12063-12063
Publication Date(Web):2012/08/03
DOI:10.1039/C2DT31425H
The reaction of the 16-electron half-sandwich complex MeCpCo(S2C2B10H10) (1b; MeCp = methylcyclopentadienyl) and methyl propiolate (HCCCO2Me) at ambient temperature leads to MeCpCo(S2C2B10H9)(CHCHCO2Me) (2), MeCpCo(S2C2B10H8)(CHCHCO2Me)2 (3), MeCpCo(S2C2B10H9)[MeO2CCCH(MeO2C)CCH)](CHCHCO2Me) (4) and MeCpCo(S2C2B10H9)(CH2CCO2Me) (5). The reaction of Me4CpCo(S2C2B10H10) (1c; Me4Cp = tetramethylcyclopentadienyl) and the alkyne gives rise to Me4CpCo(S2C2B10H10)[MeO2CCCH(MeO2C)CCH] (6) and Me4CpCo (S2C2B10H9)(CH2CCO2Me) (7). Both 2 and 3 are 16-electron complexes but containing a B(3)-substituted o-carborane-1,2-dithiolate ligand in 2 and a B(3,6)-disubstituted o-carborane-1,2-dithiolate ligand in 3, respectively. In 4 and 6, two alkynes are inserted into one Co–S bond to meet an 18 electron configuration at metal, however, 4 has one B-substitution at carborane. Both 5 and 7 have the same structural type bearing a B–CH2 unit. The reactions of Cp#Co(E2C2B10H10) [Cp# = Cp (1a), MeCp (1b), Me4Cp (1c) and Me5Cp (1d); E = S, Se] with 2-methylpropanedithioic acid (L1) or pyrrolidine-1-carbodithioic acid (L2) lead to Co[S2CCH(CH3)2]3 (8) or Co[S2CN(CH2)4]3 (9), respectively, in an octahedral geometry. The three-component reactions of 1a–1d, methyl propiolate and L1 or L2 afford seven new compounds Cp#Co[S2C2B10H10(CHCHCO2Me)][S2CCH(CH3)2] [Cp# = Cp (10a), MeCp (10b) and Me4Cp (10c)], [S2CCH(CH3)2]2Co(S2C2B10H10)(CHCCO2Me){CpCo[S2CCH(CH3)2]} (11a), Cp#Co[S2C2B10H10(CHCHCO2Me)][S2CN(CH2)4] [Cp# = Cp (12a), MeCp (12b) and Me4Cp (12c)]. All 10a–10c and 12a–12c contain one deprotonated L1 or L2 ligand and one reduced alkyne. 11a has two 18-electron Co centers linked by one reduced alkyne. One metal is coordinated by an o-carborane-1,2-dithiolate and two L1 ligands, and the other is coordinated by one L1 ligand and one η5-Cp unit. In both two- and three-component reactions the reactivity of the 16-electron half-sandwich complexes Cp#Co(S2C2B10H10) is dependent on the size of the Cp# unit. All compounds were fully characterized by spectroscopic techniques and elemental analysis. The solid-state structures of 4, 5, 7, 10a and 11a were further determined by X-ray crystallographic analysis.
Co-reporter:De-Hong Wu, Chun-Hui Wu, Yi-Zhi Li, Da-Dong Guo, Xue-Mei Wang and Hong Yan
Dalton Transactions 2009(Issue 2) pp:NaN290-290
Publication Date(Web):2008/11/10
DOI:10.1039/B810831E
The addition reactions of the 16e half-sandwich complexes (p-cymene)M(S2C2B10H10) (1S, M = Ru; 2S, M = Os) and Cp*Ir(E2C2B10H10) (3S, E = S; 3Se, E = Se) with ethynylferrocene lead selectively to the 18e complexes (p-cymene)Ru(S2C2B10H9)(H2CCFc) (Fc = ferrocenyl) (4S), (p-cymene)Os(S2C2B10H9)(H2CCFc) (5S), Cp*Ir(S2C2B10H9)(H2CCFc) (6S) and Cp*Ir(Se2C2B10H9)(H2CCFc) (6Se), in which the alkyne is regio- and stereoselectively inserted into one of the M–E bonds that may further lead to metal-induced B–H activation, hydrogen atom transfer from the carboranevia the metal center to the inserted alkyne, and the generation of a M–B bond. In all complexes the S-η2-(Fc)C–C and C–B(M) moieties occupy a cisoid position. The four new complexes are characterized by IR, MS, NMR spectroscopy and microanalysis, and the X-ray structural analysis of 4S is performed. 4S was observed to promote the uptake of anticancer drug daunorubicin in drug-resistant leukemia K562 cells.
Co-reporter:Hong-De Ye, Guan-Yu Ding, Ming-Shi Xie, Yi-Zhi Li and Hong Yan
Dalton Transactions 2011 - vol. 40(Issue 10) pp:NaN2313-2313
Publication Date(Web):2010/11/12
DOI:10.1039/C0DT00933D
The 16e half-sandwich complex Cp*Co(S2C2B10H10)(1, Cp* = pentamethylcyclopentadienyl) reacts with N-1-naphthylpropargylamide to afford two 18e complexes (2) and (3). 2 is a 1:1 adduct containing a four-membered metallacycle. In 3 the alkyne is twofold inserted into one Co–S bond in a head–head mode. Reactions of 1 with 1-(2-furyl)-2-propyn-1-one and 1-ferrocenyl-2-propyn-1-one lead to two types of alkyne twofold inserted products (4)/(5) and (6)/(7), respectively. The reaction of 1 with phenylacetylene gives rise to the sole complex (8) of the same structural type as 3, 4 and 6, whereas the reaction of 1 with dimethyl acetylene dicarboxylate affords the sole 1:1 adduct (9). Complexes 2–9 have been characterized by IR, NMR, elemental analysis and mass spectrometry, and 3–9 have also been determined by single-crystal X-ray diffraction analysis.
Co-reporter:Zhi-Wei Xu, Lei Han, Cheng Ji, Rui Zhang, Xu-Jie Shen and Hong Yan
Dalton Transactions 2011 - vol. 40(Issue 26) pp:NaN6997-6997
Publication Date(Web):2011/06/06
DOI:10.1039/C1DT10291E
The reactions of the 16e half-sandwich complex (p-cymene)Ru(S2C2B10H10) (Ru16e) with 1,4-diethynylbenzene (L1), 3′,6-diethynyl-1,1′-binaphthyl-2,7′-diyl diacetate (L2), 2-bromo-5-ethynylthiophene (L3) and 2,5-diethynylthiophene (L4) lead to 18e mononuclear complexes (p-cymene)Ru(S2C2B10H9)(H2CCPhCCH) (1), (p-cymene)Ru(S2C2B10H9)[H2CC(C24H16O4)CCH] (2), (p-cymene)Ru(S2C2B10H9) [H2CC(C4H2S)Br] (3) and (p-cymene)Ru(S2C2B10H9) [H2CC(C4H2S)CCH] (4), respectively. In all of them, metal-induced B–H activation has occurred, which leads to a stable Ru–B bond, and the structures take a cisoid arrangement. Only in the case of L4, the binuclear complexes [(p-cymene)Ru(S2C2B10H9)]2[H2CC(C4H2S)CCH2] (5a and 5b) are observed, which are conformational isomers generated by the differing orientations of the p-cymene unit. 4 can be readily converted to the complex (p-cymene)Ru(S2C2B10H9)[H2CC(C4H2S)COCH3] (6) in the presence of silica and H2O. All of these products 1–6 were characterized by NMR, IR, elemental analysis and mass spectrometry. The structures of 1, 3, and 5a were also determined by single-crystal X-ray diffraction analysis.
Co-reporter:Hongde Ye, Baohua Xu, Mingshi Xie, Yizhi Li and Hong Yan
Dalton Transactions 2011 - vol. 40(Issue 24) pp:NaN6546-6546
Publication Date(Web):2011/05/17
DOI:10.1039/C1DT10315F
The reaction of the dinuclear cobalt compound [(CpCoS2C2B10H10)(CpCoSC2B10H11)(n-C4H9S)] (1) with HCCC(O)Fc leads to the cobalt-free products (C2B10H10)(SCHCHCOFc)2 (4–6), (S2C2B10H10)(HCCCOFc) (7), and (C2B10H11)(SCHCHCOFc) (8, 9). 4–6 are produced by hydrosulfuration of the alkyne at the 1,2-dicarba-closo-dodecaborane-dithiolate ligand with the generated vinyl groups in Z/Z, Z/E and E/E configurations, respectively. In 7, the alkyne is added to 1,2-dicarba-closo-dodecaborane-dithiolate at the two sulfur sites. 8 and 9 are the products of alkyne hydrosulfuration at the 1,2-dicarba-closo-dodecaborane-1-monothiolate ligand with the generated vinyl group in either Z or E configuration. The treatment of 1 with HCCCO2Me gives rise to the parallel products (C2B10H10)(SCHCHCO2Me)2 (10–12) and (C2B10H11)(SCHCHCO2Me) (13, 14). All of the new compounds have been characterized by IR, NMR, elemental analysis and mass spectroscopy. The structures of compounds 4, 7, and 8 have also been determined by single-crystal X-ray diffraction analysis.
Co-reporter:Wei Zhong, Qibai Jiang, Qian Zhang, Yi Shang, Hong Yan and Vladimir Bregadze
Dalton Transactions 2014 - vol. 43(Issue 13) pp:NaN4968-4968
Publication Date(Web):2013/09/24
DOI:10.1039/C3DT52308J
Reactions of half-sandwich complex Cp*IrS2C2B10H10 (1) with 1-azido-3-nitrobenzene (3-NO2C6H4N3, L) upon heating or under light led to new complexes 2–6. Complexes 2 and 3 contain a five-membered cyclometalated ligand arising from C(sp2)–H activation of the azide ligand L. Complex 4 is a 16 electron species containing a new-generated C–C bond between the azide ligand L and the Cp* unit where C(sp3)–H activation of the methyl unit occurred. Complexes 5 and 6 contain two types of the ligand which appear in complexes 2, 3 and 4. Further reactions of complexes 5 and 6 with L under more harsh conditions gave rise to the nucleophilic addition products 7 and 8, where ring expansion of the azide ligand at the imido site of complexes 5 and 6 happened. Complexes 2–8 were characterized by NMR, MS, IR, and elemental analysis, and X-ray structural analyses were performed for complexes 2–4 and 6–8. The radical mechanisms for the formation of complexes 2–6 were proposed on the basis of capture experiments by EPR and ESI-MS. And the formation mechanism of complexes 7 and 8 was also suggested.
Co-reporter:Qibai Jiang, Zhaojin Wang, Yizhi Li and Hong Yan
Chemical Communications 2013 - vol. 49(Issue 52) pp:NaN5882-5882
Publication Date(Web):2013/05/13
DOI:10.1039/C3CC42554A
A 15-electron Ru(III) complex containing carborane-thiolato ligands heterolytically cleaves dihydrogen at ambient pressure and temperature to give Ru(IV)/Ru(II) species containing a hydrido ligand at Ru(IV) and SH ligand at Ru(II). In the presence of HBF4·H2O the reaction further leads to an unprecedented replacement of an S atom by an O atom in the carborane-dithiolato ligand.
Co-reporter:Chao Shi, Huibin Sun, Qibai Jiang, Qiang Zhao, Jingxia Wang, Wei Huang and Hong Yan
Chemical Communications 2013 - vol. 49(Issue 42) pp:NaN4748-4748
Publication Date(Web):2013/03/04
DOI:10.1039/C3CC40996A
Both neutral and cationic iridium(III) complexes containing carborane units were synthesized. Bulky carboranes can significantly improve phosphorescence quantum yields of these complexes and the electronic effect of carboranes can evidently tune emission wavelengths of cationic complexes.
Co-reporter:Xiang Li, Yongheng Yin, Hong Yan, Changsheng Lu and Qiang Zhao
Dalton Transactions 2017 - vol. 46(Issue 30) pp:NaN10089-10089
Publication Date(Web):2017/07/10
DOI:10.1039/C7DT02009K
A novel series of heteroleptic iridium complexes with 2-phenyl-pyridine as a main ligand and carborane-functionalized 2,2′-bipyridine as an ancillary ligand were synthesized, and characterized as [Ir(ppy)2(By)]PF6 (where ppy is 2-phenyl-pyridine, By is 5-(2-R-Cb)-2,2′-bipyridine, R = H (2a), CH3 (2b), Ph (2c), iPr (2d) and iBu (2e), or By is 4-(2-R-Cb)-2,2′-bipyridine while R = H (3a), CH3 (3b), Ph (3c), iPr (3d) and iBu (3e), Cb = o-carboran-1-yl). The R groups and the substitution sites of carborane on the pyridine ring have caused differences in the emission properties of these complexes. In addition, the quantum efficiency of [Ir(ppy)2(By)]PF6 complexes has been tuned as well through the introduction of various 2-R-substituted o-carboranes into the ancillary ligand 2,2′-bipyridine, no matter in the solid state (from 0.12 to 0.25) or in solution (from 0.04 to 0.25). The emission color was tuned from yellow to red by the o-carboranyl unit because of its inductive effect. Density functional theory (DFT) and time dependent DFT (TD-DFT) calculations have been applied to investigate excited-state electronic structures of the newly synthesized complexes, which are consistent with the observed red-shift emissions.
Co-reporter:Wei Zhong, Mingshi Xie, Qibai Jiang, Yizhi Li and Hong Yan
Chemical Communications 2012 - vol. 48(Issue 15) pp:NaN2154-2154
Publication Date(Web):2012/01/13
DOI:10.1039/C2CC16943F
The thermal or photochemical reactions of Cp*IrS2C2B10H10 (1) and aryl azides lead to C–C coupling via C(sp3)–H activation in 2 as well as the formation of C–S bonds and new-type SSN pincer ligands in 3–8 through ortho-substituted electron-withdrawing group migration over an aryl ring.
Co-reporter:Deshuang Tu, Pakkin Leong, Zhihong Li, Rongrong Hu, Chao Shi, Kenneth Yin Zhang, Hong Yan and Qiang Zhao
Chemical Communications 2016 - vol. 52(Issue 84) pp:NaN12497-12497
Publication Date(Web):2016/09/28
DOI:10.1039/C6CC07093K
An efficient strategy was designed to realize spontaneous recovery of mechanochromic luminescence by carborane-functionalized anthracene derivatives. A metastable charge-transfer emission from anthracene to o-carborane is responsible for this process.

















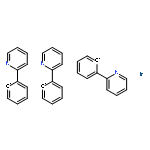
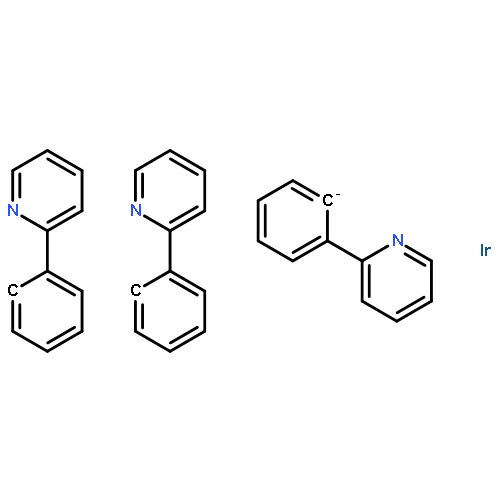





![Benzenesulfonamide, 4-[2-[[(2-hydroxyphenyl)methylene]amino]ethyl]-](http://img.cochemist.com/ccimg/320400/320337-96-0.png)
![Benzenesulfonamide, 4-[2-[[(2-hydroxyphenyl)methylene]amino]ethyl]-](http://img.cochemist.com/ccimg/320400/320337-96-0_b.png)

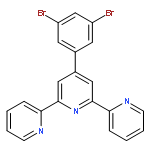
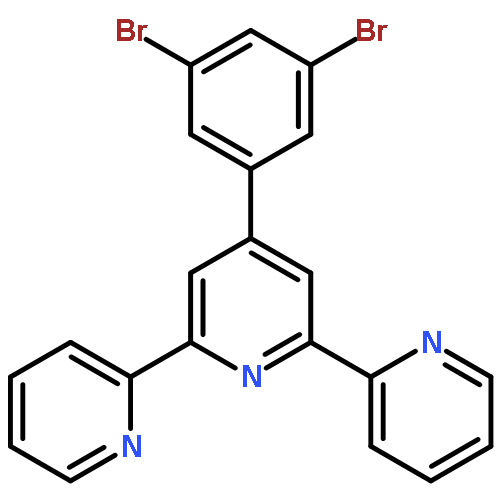












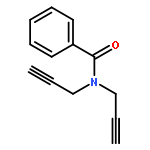
![5,7-DIHYDROXY-2-(4-METHOXYPHENYL)-8-(3-METHYLBUT-2-ENYL)-3-[(2S,3R,4R,5R,6S)-3,4,5-TRIHYDROXY-6-METHYLOXAN-2-YL]OXYCHROMEN-4-ONE](http://img.cochemist.com/ccimg/113600/113558-15-9.png)
![5,7-DIHYDROXY-2-(4-METHOXYPHENYL)-8-(3-METHYLBUT-2-ENYL)-3-[(2S,3R,4R,5R,6S)-3,4,5-TRIHYDROXY-6-METHYLOXAN-2-YL]OXYCHROMEN-4-ONE](http://img.cochemist.com/ccimg/113600/113558-15-9_b.png)
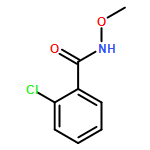












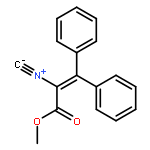
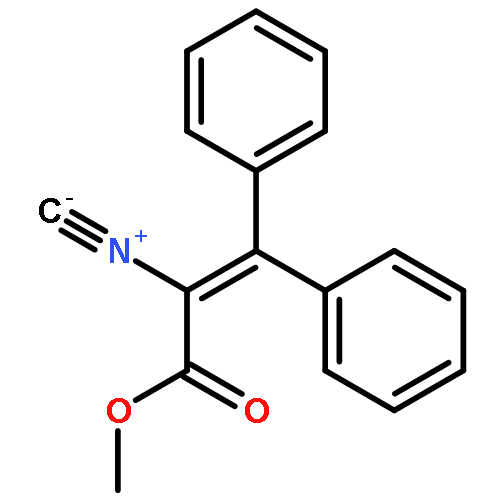
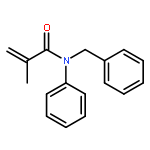
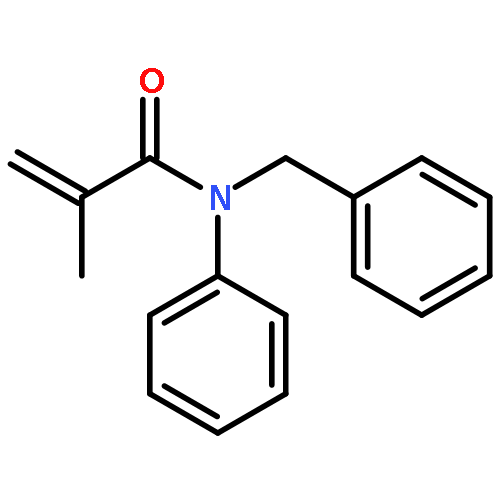
![Phenol, 2-[[[2-(4-hydroxyphenyl)ethyl]imino]methyl]-](http://img.cochemist.com/ccimg/73500/73428-20-3.png)
![Phenol, 2-[[[2-(4-hydroxyphenyl)ethyl]imino]methyl]-](http://img.cochemist.com/ccimg/73500/73428-20-3_b.png)
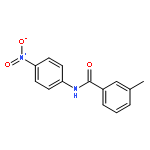

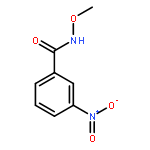
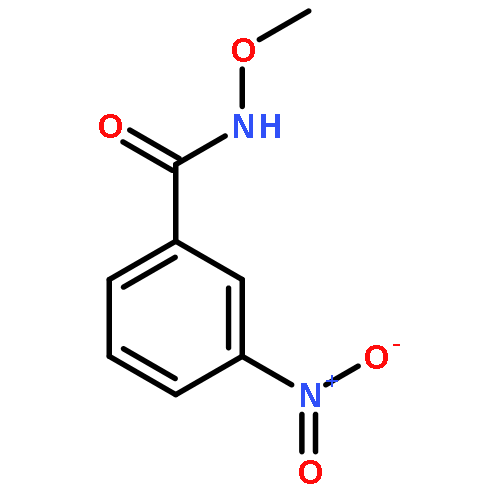



















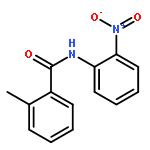
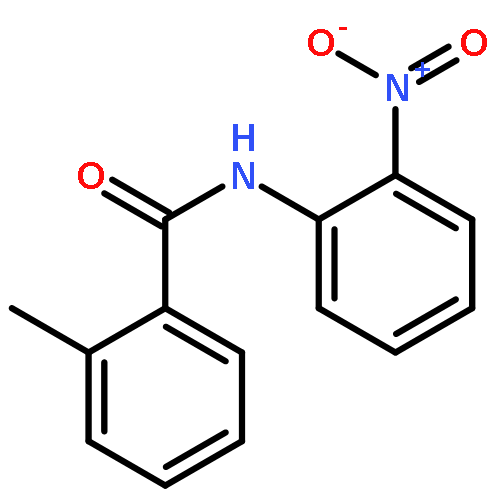
![4-Methyl-[1,1'-biphenyl]-2-amine](http://img.cochemist.com/ccimg/42400/42308-28-1.png)
![4-Methyl-[1,1'-biphenyl]-2-amine](http://img.cochemist.com/ccimg/42400/42308-28-1_b.png)


![(3aS,4S,6R,6aR)-4-(Iodomethyl)-6-methoxy-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole](http://img.cochemist.com/ccimg/38900/38838-06-1.png)
![(3aS,4S,6R,6aR)-4-(Iodomethyl)-6-methoxy-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole](http://img.cochemist.com/ccimg/38900/38838-06-1_b.png)












![Phenol, 2-[[[2-(4-morpholinyl)ethyl]imino]methyl]-](http://img.cochemist.com/ccimg/27600/27528-45-6.png)
![Phenol, 2-[[[2-(4-morpholinyl)ethyl]imino]methyl]-](http://img.cochemist.com/ccimg/27600/27528-45-6_b.png)


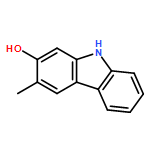
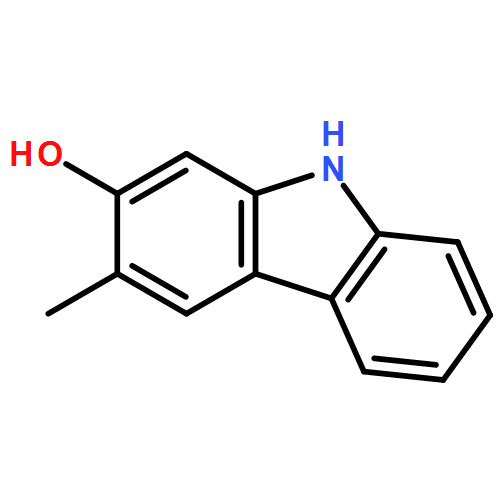


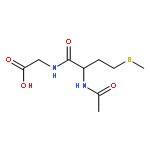

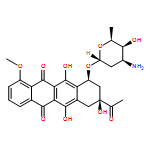













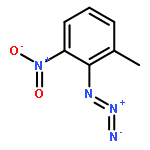












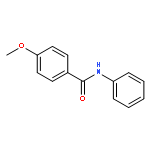
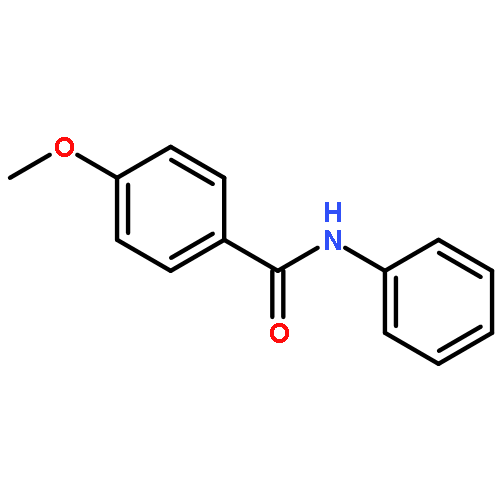
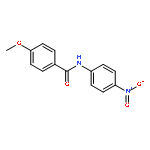
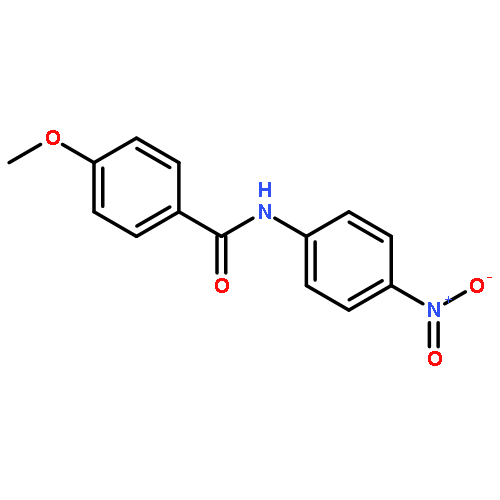


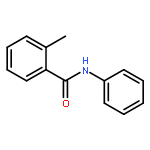




![N-[(Z)-(6-oxocyclohexa-2,4-dien-1-ylidene)methyl]glycine](http://img.cochemist.com/ccimg/6400/6343-78-8.png)
![N-[(Z)-(6-oxocyclohexa-2,4-dien-1-ylidene)methyl]glycine](http://img.cochemist.com/ccimg/6400/6343-78-8_b.png)
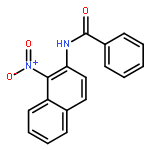
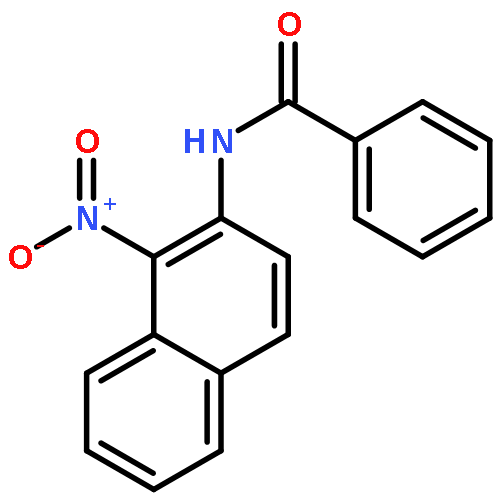





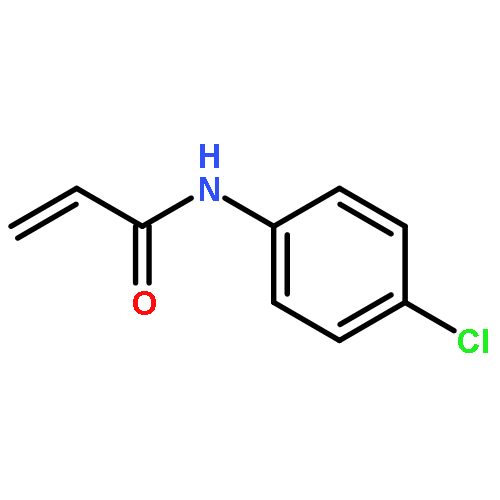






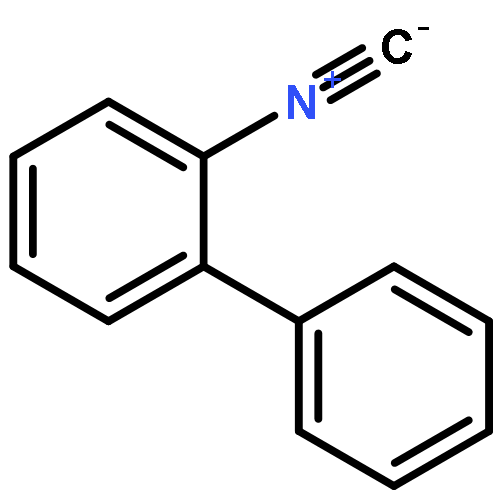






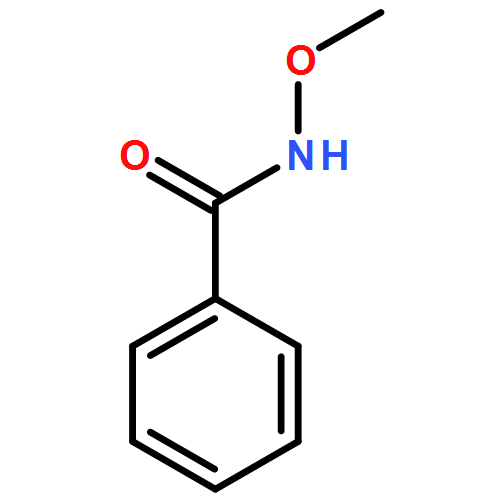




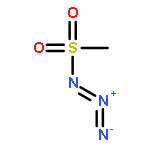
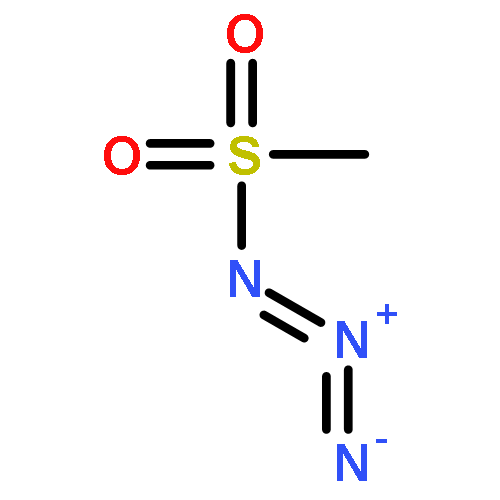
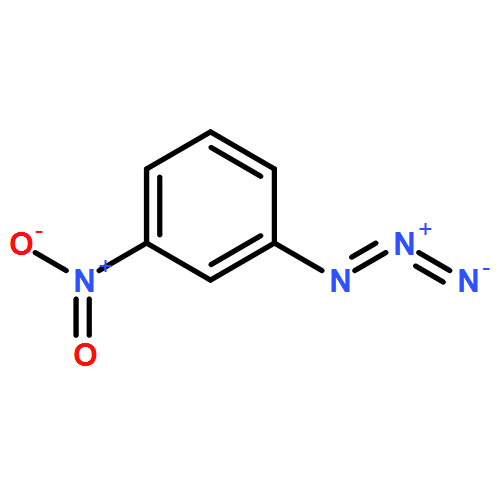


![[1,1'-Biphenyl]-2-amine, 2'-methoxy-](http://img.cochemist.com/ccimg/1300/1206-76-4.png)
![[1,1'-Biphenyl]-2-amine, 2'-methoxy-](http://img.cochemist.com/ccimg/1300/1206-76-4_b.png)












![1,6,7,8,9,15,16,17,18,18,19,19-DODECACHLOROHEXACYCLO[13.2.1.1<SUP>6,9</SUP>.0<SUP>2,14</SUP>.0<SUP>3,12</SUP>.0<SUP>5,10</SUP>]NONADECA-7,16-DIENE](http://img.cochemist.com/ccimg/400/365-33-3.png)
![1,6,7,8,9,15,16,17,18,18,19,19-DODECACHLOROHEXACYCLO[13.2.1.1<SUP>6,9</SUP>.0<SUP>2,14</SUP>.0<SUP>3,12</SUP>.0<SUP>5,10</SUP>]NONADECA-7,16-DIENE](http://img.cochemist.com/ccimg/400/365-33-3_b.png)











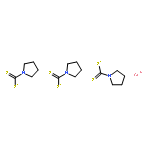
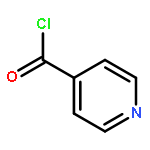
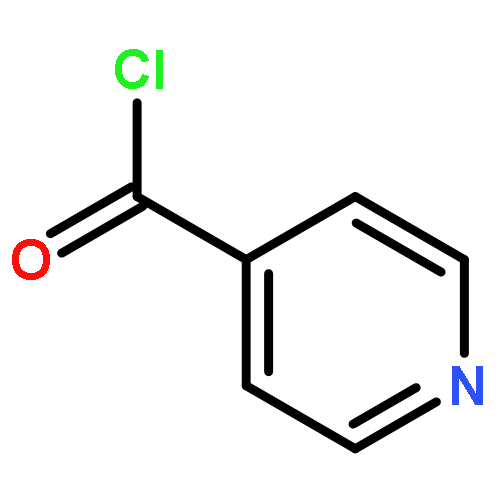






![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)