Co-reporter:Shuting Sun, Katarzyna M. Błażewska, Anastasia P. Kadina, Boris A. Kashemirov, Xuchen Duan, James T. Triffitt, James E. Dunford, R. Graham G. Russell, Frank H. Ebetino, Anke J. Roelofs, Fraser P. Coxon, Mark W. Lundy, and Charles E. McKenna
Bioconjugate Chemistry 2016 Volume 27(Issue 2) pp:329
Publication Date(Web):December 8, 2015
DOI:10.1021/acs.bioconjchem.5b00369
A bone imaging toolkit of 21 fluorescent probes with variable spectroscopic properties, bone mineral binding affinities, and antiprenylation activities has been created, including a novel linking strategy. The linking chemistry allows attachment of a diverse selection of dyes fluorescent in the visible to near-infrared range to any of the three clinically important heterocyclic bisphosphonate bone drugs (risedronate, zoledronate, and minodronate or their analogues). The resultant suite of conjugates offers multiple options to “mix and match” parent drug structure, fluorescence emission wavelength, relative bone affinity, and presence or absence of antiprenylation activity, for bone-related imaging applications.
Co-reporter:Candy S. Hwang, Alvin Kung, Boris A. Kashemirov, Chao Zhang, and Charles E. McKenna
Organic Letters 2015 Volume 17(Issue 7) pp:1624-1627
Publication Date(Web):March 17, 2015
DOI:10.1021/ol503765n
The first preparation of the individual β,γ-CHF-ATP stereoisomers 12a and 12b is reported. Configurationally differing solely by the orientation of the C–F fluorine, 12a and 12b have discrete 31P (202 MHz, pH 10.9, ΔδPα 6 Hz, ΔδPβ 4 Hz) and 19F NMR (470 MHz, pH 9.8, ΔδF 25 Hz) spectral signatures and exhibit a 6-fold difference in IC50 values for c-Src kinase, attributed to a unique interaction of the (S)-fluorine of bound 12b with R388 in the active site.
Co-reporter:Anastasia P. Kadina, Boris A. Kashemirov, Keriann Oertell, Vinod K. Batra, Samuel H. Wilson, Myron F. Goodman, and Charles E. McKenna
Organic Letters 2015 Volume 17(Issue 11) pp:2586-2589
Publication Date(Web):May 13, 2015
DOI:10.1021/acs.orglett.5b00799
Novel α,β-CH2 and β,γ-NH (1a) or α,β-NH and β,γ-CH2 (1b) “Met-Im” dTTPs were synthesized via monodemethylation of triethyl-dimethyl phosphorimido-bisphosphonate synthons (4a, 4b), formed via a base-induced [1,3]-rearrangement of precursors (3a, 3b) in a reaction with dimethyl or diethyl phosphochloridate. Anomerization during final bromotrimethylsilane (BTMS) deprotection after Mitsunobu conjugation with dT was avoided by microwave conditions. 1a was 9-fold more potent in inhibiting DNA polymerase β, attributed to an NH-group interaction with R183 in the active site.
Co-reporter:Kyle J. Seamon ; Erik C. Hansen ; Anastasia P. Kadina ; Boris A. Kashemirov ; Charles E. McKenna ; Namandjé N. Bumpus ;James T. Stivers
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:9822-9825
Publication Date(Web):July 1, 2014
DOI:10.1021/ja5035717
SAMHD1 is a GTP-activated nonspecific dNTP triphosphohydrolase that depletes dNTP pools in resting CD4+ T cells and macrophages and effectively restricts infection by HIV-1. We have designed a nonsubstrate dUTP analogue with a methylene bridge connecting the α phosphate and 5′ carbon that potently inhibits SAMHD1. Although pppCH2dU shows apparent competitive inhibition, it acts by a surprising allosteric mechanism that destabilizes active enzyme tetramer.
Co-reporter:Keriann Oertell, Brian T. Chamberlain, Yue Wu, Elena Ferri, Boris A. Kashemirov, William A. Beard, Samuel H. Wilson, Charles E. McKenna, and Myron F. Goodman
Biochemistry 2014 Volume 53(Issue 11) pp:
Publication Date(Web):March 3, 2014
DOI:10.1021/bi500101z
Kinetics studies of dNTP analogues having pyrophosphate-mimicking β,γ-pCXYp leaving groups with variable X and Y substitution reveal striking differences in the chemical transition-state energy for DNA polymerase β that depend on all aspects of base-pairing configurations, including whether the incoming dNTP is a purine or pyrimidine and if base-pairings are right (T•A and G•C) or wrong (T•G and G•T). Brønsted plots of the catalytic rate constant (log(kpol)) versus pKa4 for the leaving group exhibit linear free energy relationships (LFERs) with negative slopes ranging from −0.6 to −2.0, consistent with chemical rate-determining transition-states in which the active-site adjusts to charge-stabilization demand during chemistry depending on base-pair configuration. The Brønsted slopes as well as the intercepts differ dramatically and provide the first direct evidence that dNTP base recognition by the enzyme–primer–template complex triggers a conformational change in the catalytic region of the active-site that significantly modifies the rate-determining chemical step.
Co-reporter:Candy S. Hwang, Boris A. Kashemirov, and Charles E. McKenna
The Journal of Organic Chemistry 2014 Volume 79(Issue 11) pp:5315-5319
Publication Date(Web):May 12, 2014
DOI:10.1021/jo500452b
Jakeman et al. recently reported the inability to distinguish the diastereomers of uridine 5′-β,γ-fluoromethylenetriphosphate (β,γ-CHF-UTP, 1) by 19F NMR under conditions we previously prescribed for the resolution of the corresponding β,γ-CHF-dGTP spectra, stating further that 1 decomposed under these basic conditions. Here we show that the 19F NMR spectra of 1 (∼1:1 diastereomer mixture prepared by coupling of UMP-morpholidate with fluoromethylenebis(phosphonic acid)) in D2O at pH 10 are indeed readily distinguishable. 1 in this solution was stable for 24 h at rt.
Co-reporter:Feng Ni ; Chi Chung Lee ; Candy S. Hwang ; Yilin Hu ; Markus W. Ribbe
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10346-10352
Publication Date(Web):July 8, 2013
DOI:10.1021/ja3121058
Reduction of the first known halogen-containing substrate by nitrogenase (N2ase), 3,3-difluorocyclopropene (DFCP), was investigated. Reduction requires both N2ase proteins (MoFe and Fe protein), ATP, and an exogenous reductant (dithionite, DT), as with N2 and known alternative substrates of the enzyme. Two major products providing evidence for reductive C–F bond cleavage were confirmed, propene (P1, requiring 6e–/6H+) and 2-fluoropropene (P2, 4e–/4H+). Both were identified by GC-MS and NMR spectroscopy, and had the same Km constants (0.022 atm, 5.4 mM). Reduction of 1,2-dideuterated DFCP (d2-DFCP) further revealed that (i) in both P1 and P2, two deuterium atoms are retained, one on carbon-1 and one on carbon-3, indicating that C═C bond cleavage rather than C–C bond cleavage is involved during DFCP reduction at least to P2 (assuming no F migration); (ii) no selectivity was observed in formation of cis and trans isomers of 1,3-d2-2-fluoropropene, whereas cis-1,3-d2-propene is the predominant 1,3-d2-propene product, indicating that one of the bound reduction intermediates on the pathway to propene is constrained geometrically. A reduction mechanism, consistent with hydride transfer as a key step, is discussed. Reductive C–F bond cleavage is an ability of N2ase that further demonstrates the unique and remarkable scope of its catalytic prowess.
Co-reporter:Ivan S. Krylov, Boris A. Kashemirov, John M. Hilfinger, and Charles E. McKenna
Molecular Pharmaceutics 2013 Volume 10(Issue 2) pp:445-458
Publication Date(Web):January 22, 2013
DOI:10.1021/mp300663j
Certain acyclic nucleoside phosphonates (ANPs) such as (S)-HPMPC (cidofovir, Vistide) and (S)-HPMPA have been shown to be active against a broad spectrum of DNA and retroviruses. However, their poor absorption as well as their toxicity limit the utilization of these therapeutics in the clinic. Nucleoside phosphonates are poorly absorbed primarily due to the presence of the phosphonic acid group, which ionizes at physiological pH. When dosed intravenously they display dose-limiting nephrotoxicity due to their accumulation in the kidney. To overcome these limitations, nucleoside phosphonate prodrug strategies have taken center stage in the development pathway and a number of different approaches are at various stages of development. Our efforts have focused on the development of ANP prodrugs in which a benign amino acid promoiety masks a phosphonate P–OH via a hydroxyl side chain. The design of these prodrugs incorporates multiple chemical groups (the P–X–C linkage, the amino acid stereochemistry, the C-terminal and N-terminal functional groups) that can be tuned to modify absorption, pharmacokinetic and efficacy properties with the goal of improving overall prodrug performance.Keywords: (S)-HPMPA; (S)-HPMPC; acyclic nucleoside phosphonates; amino acid; antiviral; cidofovir; prodrugs;
Co-reporter:Yue Wu ; Valeria M. Zakharova ; Boris A. Kashemirov ; Myron F. Goodman ; Vinod K. Batra ; Samuel H. Wilson
Journal of the American Chemical Society 2012 Volume 134(Issue 21) pp:8734-8737
Publication Date(Web):March 7, 2012
DOI:10.1021/ja300218x
Deoxynucleoside 5′-triphosphate analogues in which the β,γ-bridging oxygen has been replaced with a CXY group are useful chemical probes to investigate DNA polymerase catalytic and base-selection mechanisms. A limitation of such probes has been that conventional synthetic methods generate a mixture of diastereomers when the bridging carbon substitution is nonequivalent (X ≠ Y). We report here a general solution to this long-standing problem with four examples of β,γ-CXY dNTP diastereomers: (S)- and (R)-β,γ-CHCl-dGTP (12a-1/12a-2) and (S)- and (R)-β,γ-CHF-dGTP (12b-1/12b-2). Central to their preparation was conversion of the prochiral parent bisphosphonic acids to the P,C-dimorpholinamide derivatives 7 of their (R)-mandelic acid monoesters, which provided access to the individual diastereomers 7a-1, 7a-2, 7b-1, and 7b-2 by preparative HPLC. Selective acidic hydrolysis of the P–N bond then afforded “portal” diastereomers, which were readily coupled to morpholine-activated dGMP. Removal of the chiral auxiliary by H2 (Pd/C) gave the four individual diastereomeric nucleotides 12, which were characterized by 31P, 1H, and 19F NMR spectroscopy and by mass spectrometry. After treatment with Chelex-100 to remove traces of paramagnetic ions, at pH ∼10 the diastereomer pairs 12a,b exhibit discrete Pα and Pβ31P resonances. The more upfield Pα and more downfield Pβ resonances (and also the more upfield 19F NMR resonance in 12b) are assigned to the R configuration at the Pβ-CHX-Pγ carbons on the basis of the absolute configurations of the individual diastereomers as determined from the X-ray crystallographic structures of their ternary complexes with DNA and polymerase β.
Co-reporter:Ivan S. Krylov, Valeria M. Zakharova, Michaela Serpi, Ralf Haiges, Boris A. Kashemirov, and Charles E. McKenna
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:684-689
Publication Date(Web):November 30, 2011
DOI:10.1021/jo201735f
The configuration at phosphorus in cyclic (S)-HPMPC (1, cidofovir) and (S)-HPMPA (2) phenyl ester (5 and 6, respectively) diastereomers ((Rp)-5, (Rp)-6, (Sp)-6) was determined by X-ray crystallography and correlated to their 1H and 31P NMR spectra in solution. (Rp)-5 and (Rp)-6 have chair conformations with the nucleobase substituent equatorial and the P-OPh axial. Perhaps surprisingly, (Sp)-6 is (a, a) in the crystal and exists largely as an equilibrium of (a, a)/(e, e) conformers in chloroform or acetonitrile.
Co-reporter:Brian T. Chamberlain;Dr. Vinod K. Batra;Dr. William A. Beard;Anastasia P. Kadina;David D. Shock;Dr. Boris A. Kashemirov;Dr. Charles E. McKenna;Dr. Myron F. Goodman;Dr. Samuel H. Wilson
ChemBioChem 2012 Volume 13( Issue 4) pp:528-530
Publication Date(Web):
DOI:10.1002/cbic.201100738
Co-reporter:Katarzyna M. Błażewska, Ralf Haiges, Boris A. Kashemirov, Frank H. Ebetino and Charles E. McKenna
Chemical Communications 2011 vol. 47(Issue 22) pp:6395-6397
Publication Date(Web):09 May 2011
DOI:10.1039/C1CC10876J
Under certain conditions, the phosphonocarboxylate analogue (3) of the bisphosphonate drug minodronate (4) in contact with borosilicate glassware reversibly forms an isolable dimer complex of boron, as revealed by the X-ray crystallographic structure of the (R,R/S,S) complex and supported by NMR and HRMS data.
Co-reporter:Valeria M. Zakharova ; Michaela Serpi ; Ivan S. Krylov ; Larryn W. Peterson ; Julie M. Breitenbach ; Katherine Z. Borysko ; John C. Drach ; Mindy Collins ; John M. Hilfinger ; Boris A. Kashemirov
Journal of Medicinal Chemistry 2011 Volume 54(Issue 16) pp:5680-5693
Publication Date(Web):August 3, 2011
DOI:10.1021/jm2001426
Eight novel single amino acid (6–11) and dipeptide (12, 13) tyrosine P–O esters of cyclic cidofovir ((S)-cHPMPC, 4) and its cyclic adenine analogue ((S)-cHPMPA, 3) were synthesized and evaluated as prodrugs. In vitro IC50 values for the prodrugs (<0.1−50 μM) vs vaccinia, cowpox, human cytomegalovirus, and herpes simplex type 1 virus were compared to those for the parent drugs ((S)-HPMPC, 2; (S)-HPMPA, 1; IC50 0.3–35 μM); there was no cytoxicity with KB or HFF cells at ≤100 μM. The prodrugs exhibited a wide range of half-lives in rat intestinal homogenate at pH 6.5 (<30–1732 min) with differences of 3–10× between phostonate diastereomers. The tyrosine alkylamide derivatives of 3 and 4 were the most stable. (l)-Tyr-NH-i-Bu cHPMPA (11) was converted in rat or mouse plasma solely to two active metabolites and had significantly enhanced oral bioavailability vs parent drug 1 in a mouse model (39% vs <5%).
Co-reporter:Katarzyna M. Błażewska, Feng Ni, Ralf Haiges, Boris A. Kashemirov, Fraser P. Coxon, Charlotte A. Stewart, Rudi Baron, Michael J. Rogers, Miguel C. Seabra, Frank H. Ebetino, Charles E. McKenna
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:4820-4826
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.04.063
Phosphonocarboxylate (PC) analogues of bisphosphonates are of interest due to their selective inhibition of a key enzyme in the mevalonate pathway, Rab geranylgeranyl transferase (RGGT). The dextrarotatory enantiomer of 2-hydroxy-3-(imidazo[1,2-a]pyridin-3-yl)-2-phosphonopropanoic acid (3-IPEHPC, 1) is the most potent PC-type RGGT inhibitor thus far identified. The absolute configuration of (+)-1 in the active site complex has remained unknown due to difficulties in obtaining RGGT inhibitor complex crystals suitable for X-ray diffraction analysis. However, we have now succeeded in crystallizing (−)-1 and here report its absolute configuration (AC) obtained by X-ray crystallography, thus also defining the AC of (+)-1. An Autodock Vina 1.1 computer modeling study of (+)-1 in the active site of modified RGGT binding GGPP (3DSV) identifies stereochemistry-dependent interactions that could account for the potency of (+)-1 and supports the hypothesis that this type of inhibitor binds at the TAG tunnel, inhibiting the second geranylgeranylation step. We also report a convenient 31P NMR method to determine enantiomeric excess of 1 and its pyridyl analogue 2, using α- and β-cyclodextrins as chiral solvating agents, and describe the synthesis of a small series of 1 α-X (X = H, F, Cl, Br; 7a–d) analogues to assess the contribution of the α-OH group to activity at enzyme and cellular levels. The IC50 of 1 was 5–10× lower than 7a–d, and the LED for inhibition of Rab11 prenylation in vitro was 2–8× lower than for 7a–d. However, in a viability reduction assay with J774 cells, 1 and 7b had similar IC50 values, ∼10× lower than those of 7a and 7c–d.Highlights► The absolute configuration of (+)-3-IPEHPC ((+)-1) is identified. ► The stereospecificity of (+)-1 binding to RGGTase is explained. ► α-Desoxy and α-halo analogues (7a–d) of (+)-1 are less potent. ► A convenient NMR determination of ee in 1 and 7a–d is described.
Co-reporter:Larryn W. Peterson, Jae-Seung Kim, Paul Kijek, Stefanie Mitchell, John Hilfinger, Julie Breitenbach, Kathy Borysko, John C. Drach, Boris A. Kashemirov, Charles E. McKenna
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 13) pp:4045-4049
Publication Date(Web):1 July 2011
DOI:10.1016/j.bmcl.2011.04.126
We report the synthesis and biological evaluation of Ala–(Val–)l-Ser–CO2R prodrugs of 1, where a dipeptide promoiety is conjugated to the P(OH)2 group of cidofovir (1) via esterification by the Ser side chain hydroxyl group and an ethyl group (4 and 5) or alone (6 and 7). In a murine model, oral administration of 4 or 5 did not significantly increase total cidofovir species in the plasma compared to 1 or 2, but 7 resulted in a 15-fold increase in a rat model and had an in vitro EC50 value against human cytomegalovirus comparable to 1. Neither 6 nor 7 exhibited toxicity up to 100 μM in KB or HFF cells.
Co-reporter:Dana A. Mustafa, Boris A. Kashemirov, Charles E. McKenna
Tetrahedron Letters 2011 Volume 52(Issue 18) pp:2285-2287
Publication Date(Web):4 May 2011
DOI:10.1016/j.tetlet.2011.02.058
A simple, rapid one-pot synthesis of heterocyclic (risedronate, zoledronate) and aminoalkyl (pamidronate, alendronate, neridronate) bisphosphonate drugs using microwave irradiation is presented. Good yields of product are achieved within 20 min, in contrast to conventional synthesis, which typically requires a day or longer.
Co-reporter:Brian T. Chamberlain, Thomas G. Upton, Boris A. Kashemirov, and Charles E. McKenna
The Journal of Organic Chemistry 2011 Volume 76(Issue 12) pp:5132-5136
Publication Date(Web):April 4, 2011
DOI:10.1021/jo200045a
The first examples of α-azido bisphosphonates [(RO)2P(O)]2CXN3 (1, R = i-Pr, X = Me; 2, R = i-Pr, X = H; 3, R = H, X = Me; 4, R = H, X = H) and corresponding β,γ-CXN3 dGTP (5–6) and α,β-CXN3 dATP (7–8) analogues are described. The individual diastereomers of 7 (7a/b) were obtained by HPLC separation of the dADP synthetic precursor (14a/b).
Co-reporter:Vinod K. Batra ; Lars C. Pedersen ; William A. Beard ; Samuel H. Wilson ; Boris A. Kashemirov ; Thomas G. Upton ; Myron F. Goodman
Journal of the American Chemical Society 2010 Volume 132(Issue 22) pp:7617-7625
Publication Date(Web):May 13, 2010
DOI:10.1021/ja909370k
β,γ-Fluoromethylene analogues of nucleotides are considered to be useful mimics of the natural substrates, but direct structural evidence defining their active site interactions has not been available, including the influence of the new chiral center introduced at the CHF carbon, as in β,γ-fluoromethylene-dGTP, which forms an active site complex with DNA polymerase β, a repair enzyme that plays an important role in base excision repair (BER) and oncogenesis. We report X-ray crystallographic results for a series of β,γ-CXY dGTP analogues, where X,Y = H, F, Cl, Br, and/or CH3. For all three R/S monofluorinated analogues examined (CHF, 3/4; CCH3F, 13/14; CClF 15/16), a single CXF-diastereomer (3, 13, 16) is observed in the active site complex, with the CXF fluorine atom at a ∼3 Å (bonding) distance to a guanidinium N of Arg183. In contrast, for the CHCl, CHBr, and CHCH3 analogues, both diasteromers (6/7, 8/9, 10/11) populate the dGTP site in the enzyme complex about equally. The structures of the bound dichloro (5) and dimethyl (12) analogue complexes indicate little to no steric effect on the placement of the bound nucleotide backbone. The results suggest that introduction of a single fluorine atom at the β,γ-bridging carbon atom of these dNTP analogues enables a new, stereospecific interaction within the preorganized active site complex that is unique to fluorine. The results also provide the first diverse structural data set permitting an assessment of how closely this class of dNTP analogues mimics the conformation of the parent nucleotide within the active site complex.
Co-reporter:Charles E. McKenna ; Boris A. Kashemirov ; Katarzyna M. Błażewska ; Isabelle Mallard-Favier ; Charlotte A. Stewart ; Javier Rojas ; Mark W. Lundy ; Frank H. Ebetino ; Rudi A. Baron ; James E. Dunford ; Marie L. Kirsten ; Miguel C. Seabra ; Joy L. Bala ; Mong S. Marma ; Michael J. Rogers ;Fraser P. Coxon
Journal of Medicinal Chemistry 2010 Volume 53(Issue 9) pp:3454-3464
Publication Date(Web):April 15, 2010
DOI:10.1021/jm900232u
3-(3-Pyridyl)-2-hydroxy-2-phosphonopropanoic acid (3-PEHPC, 1) is a phosphonocarboxylate (PC) analogue of 2-(3-pyridyl)-1-hydroxyethylidenebis(phosphonic acid) (risedronic acid, 2), an osteoporosis drug that decreases bone resorption by inhibiting farnesyl pyrophosphate synthase (FPPS) in osteoclasts, preventing protein prenylation. 1 has lower bone affinity than 2 and weakly inhibits Rab geranylgeranyl transferase (RGGT), selectively preventing prenylation of Rab GTPases. We report here the synthesis and biological studies of 2-hydroxy-3-imidazo[1,2-a]pyridin-3-yl-2-phosphonopropionic acid (3-IPEHPC, 3), the PC analogue of minodronic acid 4. Like 1, 3 selectively inhibited Rab11 vs. Rap 1A prenylation in J774 cells, and decreased cell viability, but was 33−60× more active in these assays. After resolving 3 by chiral HPLC (>98% ee), we found that (+)-3-E1 was much more potent than (−)-3-E2 in an isolated RGGT inhibition assay, ∼17× more potent (LED 3 μM) than (−)-3-E2 in inhibiting Rab prenylation in J774 cells and >26× more active in the cell viability assay. The enantiomers of 1 exhibited a 4-fold or smaller potency difference in the RGGT and prenylation inhibition assays.
Co-reporter:Larryn W. Peterson, Monica Sala-Rabanal, Ivan S. Krylov, Michaela Serpi, Boris A. Kashemirov, and Charles E. McKenna
Molecular Pharmaceutics 2010 Volume 7(Issue 6) pp:2349-2361
Publication Date(Web):October 7, 2010
DOI:10.1021/mp100186b
Cidofovir (HPMPC), a broad spectrum antiviral agent, cannot be administered orally due to ionization of its phosphonic acid group at physiological pH. One prodrug approach involves conversion to the cyclic form (cHPMPC, 1) and esterification by the side chain hydroxyl group of a peptidomimetic serine. Transport studies in a rat model have shown enhanced levels of total cidofovir species in the plasma after oral dosing with l-Val-l-Ser-OMe cHPMPC, 2a. To explore the possibility that 2a and its three l/d stereoisomers 2b−d undergo active transport mediated by the peptide-specific intestinal transporter PEPT1, we performed radiotracer uptake and electrophysiology experiments applying the two-electrode voltage clamp technique in Xenopus laevis oocytes overexpressing human PEPT1 (hPEPT1, SLC15A1). 2a−d did not induce inward currents, indicating that they are not transported, but the stereoisomers with an l-configuration at the N-terminal valine (2a and 2b) potently inhibited transport of the hPEPT1 substrate glycylsarcosine (Gly-Sar). A “reversed” dipeptide conjugate, l-Ser-l-Ala-OiPr cHPMPC (4), also did not exhibit detectable transport, but completely abolished the Gly-Sar signal, suggesting that affinity of the transporter for these prodrugs is not impaired by a proximate linkage to the drug in the N-terminal amino acid of the dipeptide. Single amino acid conjugates of cHPMPC (3a and 3b) or cHPMPA (5, 6a and 6b) were not transported and only weakly inhibited Gly-Sar transport. The known hPEPT1 prodrug substrate valacyclovir (7) and its l-Val-l-Val dipeptide analogue (8) were used to verify coupled transport by the oocyte model. The results indicate that the previously observed enhanced oral bioavailability of 2a relative to the parent drug is unlikely to be due to active transport by hPEPT1. Syntheses of the novel compounds 2b−d and 3−6 are described, including a convenient solid-phase method to prepare 5, 6a and 6b.Keywords: hPEPT1 transporter; intestinal transport; oral bioavailability; prodrugs; SLC15A1; Xenopus laevis oocyte;
Co-reporter:Jian-Jung Pan, Boris A. Kashemirov, Joanne Lee, Charles E. McKenna, Frank J. Devlin, Philip J. Stephens
Bioorganic Chemistry 2010 Volume 38(Issue 1) pp:7-16
Publication Date(Web):February 2010
DOI:10.1016/j.bioorg.2009.02.001
Phosphoryl-transfer reactions have long been of interest due to their importance in maintaining numerous cellular functions. A phosphoryl-transfer reaction results in two possible stereochemical outcomes: either retention or inversion of configuration at the transferred phosphorus atom. When the product is phosphate, isotopically-labeled [16O, 17O, 18O]-phosphate derivatives can be used to distinguish these outcomes; one oxygen must be replaced by sulfur or esterified to achieve isotopic chirality. Conventionally, stereochemical analysis of isotopically chiral phosphate has been based on 31P NMR spectroscopy and involves complex chemical or enzymatic transformations. An attractive alternative would be direct determination of the enantiomeric excess using chiroptical spectroscopy. (S)-Methyl-[16O, 17O, 18O]-phosphate (MePi∗), 7 and enantiomeric [16O, 17O, 18O]-thiophosphate (TPi∗), 10, were previously reported to exhibit weak electronic circular dichroism (ECD), although with 10 the result was considered to be uncertain. We have now re-examined the possibility that excesses of 7 and 10 enantiomers can be detected by ECD spectrometry, using both experimental and theoretical approaches. 7 and both the (R) and (S) enantiomers of 10 (10a, 10b) were synthesized by the ‘Oxford route’ and characterized by 1H, 31P and 17O NMR, and by MS analysis. Weak ECD could be found for 7, with suboptimal S/N. No significant ECD could be detected for the 10 enantiomers.Time-dependent DFT (TDDFT) calculations of the electronic excitation energies and rotational strengths of the same three enantiomers were carried out using the functional B3LYP and the basis set 6-311G∗∗. The isotopically-perturbed geometries were predicted using the anharmonic vibrational frequency calculational code in GAUSSIAN 03. In the case of 10, calculations were also carried out for the hexahydrated complex to investigate the influence of the aqueous solvent. The predicted excitation wavelengths are greater than the observed wavelengths, a not unusual result of TDDFT calculations. The predicted anisotropy ratios are 2.9 × 10−5 for 7, −5.3 × 10−6 for 10a/b, and 1.7 × 10−6 for 10a/b⋅(H2O)6. For 7 the predicted anisotropy ratio approximates that observed in this work, 4.5 × 10−5 at 208 nm. For 10a/b, the upper limits of the experimental anisotropy ratios (<5 × 10−6 at 225 nm, pH 9; <5 × 10−6 at 236 nm, pH 12) are comparable to the predicted magnitude of the value for 10a/b. The lower predicted value for 10a/b · (H2O)6 suggests that the aqueous environment affects the ECD significantly. Altogether, the TDDFT calculations together with a stereochemical analysis based on NMR and the MS data support the conclusion that the experimental ECD results for MePi∗ and TPi∗ may be reliable in order of magnitude.
Co-reporter:Charles E. McKenna, Boris A. Kashemirov, Larryn W. Peterson, Myron F. Goodman
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2010 Volume 1804(Issue 5) pp:1223-1230
Publication Date(Web):May 2010
DOI:10.1016/j.bbapap.2010.01.005
Abnormal replication of DNA is associated with many important human diseases, most notably viral infections and neoplasms. Existing approaches to chemotherapeutics for diseases associated with dysfunctional DNA replication classically involve nucleoside analogues that inhibit polymerase activity due to modification in the nucleobase and/or ribose moieties. These compounds must undergo multiple phosphorylation steps in vivo, converting them into triphosphosphates, in order to inhibit their targeted DNA polymerase. Nucleotide monophosphonates enable bypassing the initial phosphorylation step at the cost of decreased bioavailability. Relatively little attention has been paid to higher nucleotides (corresponding to the natural di- and triphosphate DNA polymerase substrates) as drug platforms due to their expected poor deliverability. However, a better understanding of DNA polymerase mechanism and fidelity dependence on the triphosphate moiety is beginning to emerge, aided by systematic incorporation into this group of substituted methylenebisphosphonate probes. Meanwhile, other bridging, as well as non-bridging, modifications have revealed intriguing possibilities for new drug design. We briefly survey some of this recent work, and argue that the potential of nucleotide-based drugs, and intriguing preliminary progress in this area, warrant acceptance of the challenges that they present with respect to bioavailability and metabolic stability.
Co-reporter:Thomas G. Upton, Boris A. Kashemirov, Charles E. McKenna, Myron F. Goodman, G. K. Surya Prakash, Roman Kultyshev, Vinod K. Batra, David D. Shock, Lars C. Pedersen, William A. Beard and Samuel H. Wilson
Organic Letters 2009 Volume 11(Issue 9) pp:1883-1886
Publication Date(Web):April 7, 2009
DOI:10.1021/ol701755k
α,β-Difluoromethylene deoxynucleoside 5′-triphosphates (dNTPs, N = A or C) are advantageously obtained via phosphorylation of corresponding dNDP analogues using catalytic ATP, PEP, nucleoside diphosphate kinase, and pyruvate kinase. DNA pol β Kd values for the α,β-CF2 and unmodified dNTPs, α,β-NH dUTP, and the α,β-CH2 analogues of dATP and dGTP are discussed in relation to the conformations of α,β-CF2 dTTP versus α,β-NH dUTP bound into the enzyme active site.
Co-reporter:Ernestas Gaidamauskas, Helen Parker, Boris A. Kashemirov, Alvin A. Holder, Kanokkarn Saejueng, Charles E. McKenna, Debbie C. Crans
Journal of Inorganic Biochemistry 2009 Volume 103(Issue 12) pp:1652-1657
Publication Date(Web):December 2009
DOI:10.1016/j.jinorgbio.2009.09.006
The coordination chemistry of bisphosphonates with Yb3+ was investigated to evaluate the potential of the UV–vis based detection method using the Yb3+–pyrocatechol complexation reaction as a sensor for bisphosphonates. The complexation chemistry of Yb3+ with phosphate and ATP analogs was previously described (E. Gaidamauskas, K. Saejueng, A.A. Holder, S. Bharuah, B.A. Kashemirov, D.C. Crans, C.E. McKenna, J. Biol. Inorg. Chem. 13 (2008) 1291–1299), and we here studied the complexation chemistry of bisphosphonates in this system. The spectrophotometric assay yields direct evidence for formation of a 4:3 metal to ligand complex at neutral pH. Direct evidence for Yb3+:methylenebis(phosphonate) complexes with 1:1 and 1:2 stoichiometry was also obtained by potentiometry at acidic and basic pH. Direct evidence for complex formation was obtained using 1H NMR spectroscopy although the stoichiometry was not accessed at neutral pH. Our results suggest that the spectroscopic observation of the YbPV complex can be used to conveniently measure concentrations of bisphosphonates down to 2–3 μM.
Co-reporter:Boris A. Kashemirov, Joy Lynn F. Bala, Xiaolan Chen, F. H. Ebetino, Zhidao Xia, R. Graham G. Russell, Fraser P. Coxon, Anke J. Roelofs, Michael J. Rogers and Charles E. McKenna
Bioconjugate Chemistry 2008 Volume 19(Issue 12) pp:2308
Publication Date(Web):November 25, 2008
DOI:10.1021/bc800369c
We report synthesis of the first fluorescently labeled conjugates of risedronate (1), using an epoxide linker strategy enabling conjugation of 1 via its pyridyl nitrogen with the label (carboxyfluorescein). Unlike prior approaches to create fluorescent bisphosphonate probes, the new linking chemistry did not abolish the ability to inhibit protein prenylation in vitro, while significantly retaining hydroxyapatite affinity. The utility of a fluorescent 1 conjugate in visualizing osteoclast resorption in vitro was demonstrated.
Co-reporter:Ulrika Eriksson, Larryn W. Peterson, Boris A. Kashemirov, John M. Hilfinger, John C. Drach, Katherine Z. Borysko, Julie M. Breitenbach, Jae Seung Kim, Stefanie Mitchell, Paul Kijek and Charles E. McKenna
Molecular Pharmaceutics 2008 Volume 5(Issue 4) pp:598-609
Publication Date(Web):May 16, 2008
DOI:10.1021/mp8000099
Cidofovir (HPMPC, 1), a broad-spectrum antiviral agent, is currently used to treat AIDS-related human cytomegalovirus (HCMV) retinitis and has recognized therapeutic potential for orthopox virus infections, but is limited by its low oral bioavailability. Cyclic cidofovir (2) displays decreased nephrotoxicity compared to 1, while also exhibiting potent antiviral activity. Here we describe in detail the synthesis and evaluation as prodrugs of four cHPMPC dipeptide conjugates in which the free POH of 2 is esterified by the Ser side chain alcohol group of an X-l-Ser(OMe) dipeptide: 3 (X = l-Ala), 4 (X = l-Val), 5 (X = l-Leu), and 6 (X = l-Phe). Perfusion studies in the rat establish that the mesenteric permeability to 4 is more than 20-fold greater than to 1, and the bioavailability of 4 is increased 6-fold relative to 1 in an in vivo murine model. In gastrointestinal and liver homogenates, the cHPMPC prodrugs are rapidly hydrolyzed to 2. Prodrugs 3, 4, and 5 are nontoxic at 100 μM in HFF and KB cells and in cell-based plaque reduction assays had IC50 values of 0.1−0.5 μM for HCMV and 10 μM for two orthopox viruses (vaccinia and cowpox). The enhanced transport properties of 3−6, conferred by incorporation of a biologically benign dipeptide moiety, and the facile cleavage of the Ser−O−P linkage suggest that these prodrugs represent a promising new approach to enhancing the bioavailability of 2. Keywords: antiviral activity; Cyclic cidofovir; drug delivery; oral bioavailability; prodrug;
Co-reporter:Sylvine Deprèle, Boris A. Kashemirov, James M. Hogan, Frank H. Ebetino, Bobby L. Barnett, Artem Evdokimov, Charles E. McKenna
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 9) pp:2878-2882
Publication Date(Web):1 May 2008
DOI:10.1016/j.bmcl.2008.03.088
The complex formed from crystallization of human farnesyl pyrophosphate synthase (hFPPS) from a solution of racemic [6,7-dihydro-5H-cyclopenta[c]pyridin-7-yl(hydroxy)methylene]bis(phosphonic acid) (NE-10501, 8), a chiral analog of the anti-osteoporotic drug risedronate, contained the R enantiomer in the enzyme active site. This enantiospecificity was assessed by computer modeling of inhibitor–active site interactions using Autodock 3, which was also evaluated for predictive ability in calculations of the known configurations of risedronate, zoledronate, and minodronate complexed in the active site of hFPPS. In comparison with these structures, the 8 complex exhibited certain differences, including the presence of only one Mg2+, which could contribute to its 100-fold higher IC50. An improved synthesis of 8 is described, which decreases the number of steps from 12 to 8 and increases the overall yield by 17-fold.Racemic 8, synthesized by an improved method, is preferentially bound to its inhibitor target, FPPS, as the (R) enantiomer (X-ray structure), confirming a stereospecific interaction predicted by a docking simulation.
Co-reporter:Ernestas Gaidamauskas;Kanokkarn Saejueng
JBIC Journal of Biological Inorganic Chemistry 2008 Volume 13( Issue 8) pp:1291-1299
Publication Date(Web):2008 November
DOI:10.1007/s00775-008-0415-2
Metal complexation reactions can be used effectively as sensors to measure concentrations of phosphate and phosphate analogs. Recently, a method was described for the detection of phosphate or ATP in aqueous solution based on the displacement by these ligands of pyrocatechol violet (PV) from a putative 2:1 (Yb3+)2PV complex. We have not been able to reproduce this stoichiometry and report this work in order to correct the coordination chemistry upon which sensor applications are based. In our work, colorimetric and spectrophotometric detection of phosphate was confirmed qualitatively (blue PV + Yb3+; yellow + Pi); however, the sequence of visual changes on the titration of PV with 2 equiv. of Yb3+ and back titration with ATP as described previously could not be reproduced. In contrast to the linear response to Pi that was reported previously, the absorbance response at 443 or 623 nm was found to be sigmoidal using the recommended 2:1 Yb3+:PV solution (100 μM:50 μM, pH 7, HEPES). Furthermore, both continuous variation titration and molar ratio analysis (Job plot) experiments are consistent with 1:1, not 2:1, YbPV complex stoichiometry at pH 7 in HEPES buffer, indicating that the deviation from linearity is produced by excess Yb3+. Indeed, using a 1:1 Yb3+:PV ratio produces a linear response in ΔAbs at 443 or 623 nm on back titration with analyte (phosphate or ATP). In addition, speciation analysis of the Yb–ATP system demonstrates that a 1:1 complex containing Yb3+ and ATP predominates in solution at μM metal ion and ATP concentrations. Paramagnetic 1H NMR spectroscopy directly establishes the formation of Yb3+–solute complexes in dilute aqueous solution. The 1:1 YbPV complex can be used for the colorimetric measurement of phosphate and ATP concentrations from ~2 μM.
Co-reporter:Ulrika Eriksson, John M. Hilfinger, Jae-Seung Kim, Stefanie Mitchell, Paul Kijek, Katherine Z. Borysko, Julie M. Breitenbach, John C. Drach, Boris A. Kashemirov, Charles E. McKenna
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 3) pp:583-586
Publication Date(Web):1 February 2007
DOI:10.1016/j.bmcl.2006.11.012
Cidofovir (HPMPC) is a broad-spectrum anti-viral agent whose potential, particularly in biodefense scenarios, is limited by its low oral bioavailability. Two prodrugs (3 and 4) created by conjugating ethylene glycol-linked amino acids (L-Val, L-Phe) with the cyclic form of cidofovir (cHPMPC) via a P–O ester bond were synthesized and their pH-dependent stability (3 and 4), potential for in vivo reconversion to drug (3), and oral bioavailability (3) were evaluated. The prodrugs were stable in buffer between pH 3 and 5, but underwent rapid hydrolysis in liver (t1/2 = 3.7 min), intestinal (t1/2 = 12.5 min), and Caco-2 cell homogenates (t1/2 = 20.2 min). In vivo (rat), prodrug 3 was >90% reconverted to cHPMPC. The prodrug was 4× more active than ganciclovir (IC50 value, 0.68 μM vs 3.0 μM) in a HCMV plaque reduction assay. However, its oral bioavailability in a rat model was similar to the parent drug. The contrast between the promising activation properties and unenhanced transport of the prodrug is briefly discussed.The synthesis, biological activation and oral bioavailability of an ethylene glycol-linked L-valyl ester conjugate of cyclic cidofovir (3) is reported.
Co-reporter:Charles E. McKenna, Boris A. Kashemirov, Ulrika Eriksson, Gordon L. Amidon, Phillip E. Kish, Stefanie Mitchell, Jae-Seung Kim, John M. Hilfinger
Journal of Organometallic Chemistry 2005 Volume 690(Issue 10) pp:2673-2678
Publication Date(Web):16 May 2005
DOI:10.1016/j.jorganchem.2005.03.004
Cidofovir (HPMPC, Vistide®) is a broad-spectrum anti-viral agent that is used to treat AIDS-related CMV retinitis. Currently, cidofovir is of particular interest as a potential therapy for orthopox virus infections, including smallpox. An important limitation of cidofovir and analogous nucleotide drugs in a therapeutic role is their low oral bioavailability and poor transport into cells. In principle, bioavailability of a drug can be improved by structural modification targeting transporters expressed in human intestine. To be effective, the transported prodrug must be cleaved by endogenous enzymes to its parent compound. In this work, three examples of novel cyclic cidofovir (cHPMPC) prodrugs incorporating dipeptides were synthesized and evaluated in a rat oral bioavailability model, in which the prodrugs showed significantly enhanced transport vs. HPMPC and cHPMPC. The prodrugs inhibited Gly–Sar uptake in a competitive binding assay using DC5 cells over-expressing hPepT1.Cidofovir (HPMPC, Vistide®) is a broad-spectrum anti-viral agent that is used to treat AIDS-related CMV retinitis. Currently, cidofovir is of particular interest as a potential therapy for orthopox virus infections, including smallpox. An important limitation of cidofovir and analogous nucleotide drugs in a therapeutic role is their low oral bioavailability and poor transport into cells. In principle, bioavailability of a drug can be improved by structural modification targeting transporters expressed in human intestine. To be effective, the transported prodrug must be cleaved by endogenous enzymes to its parent compound. In this work, three examples of novel cyclic cidofovir (cHPMPC) prodrugs incorporating dipeptides were synthesized and evaluated in a rat oral bioavailability model, in which the prodrugs showed significantly enhanced transport vs. HPMPC and cHPMPC. The prodrugs inhibited Gly–Sar uptake in a competitive binding assay using DC5 cells over-expressing hPepT1.
Co-reporter:Jenny A.F. Vermeer, Ineke D.C. Jansen, Matangi Marthi, Fraser P. Coxon, Charles E. McKenna, Shuting Sun, Teun J. de Vries, Vincent Everts
Bone (November 2013) Volume 57(Issue 1) pp:
Publication Date(Web):1 November 2013
DOI:10.1016/j.bone.2013.08.007
•Long-bone and jaw osteoclast precursors respond differently to bisphosphonates.•Jaw osteoclast precursors internalize more bisphosphonate than long-bone precursors.•More unprenylated Rap1a accumulates in bisphosphonate-treated jaw osteoclast precursors than in those from long bone.•Despite of different effects on prenylation, bisphosphonates similarly affect long-bone and jaw osteoclastogenesis.Bisphosphonates (BPs) are widely used in the treatment of several bone diseases, such as osteoporosis and cancers that have metastasized to bone, by virtue of their ability to inhibit osteoclastic bone resorption. Previously, it was shown that osteoclasts present at different bone sites have different characteristics. We hypothesized that BPs could have distinct effects on different populations of osteoclasts and their precursors, for example as a result of a different capacity to endocytose the drugs. To investigate this, bone marrow cells were isolated from jaw and long bone from mice and the cells were primed to differentiate into osteoclasts with the cytokines M-CSF and RANKL. Before fusion occurred, cells were incubated with fluorescein-risedronate (FAM-RIS) for 4 or 24 h and uptake was determined by flow cytometry. We found that cultures obtained from the jaw internalized 1.7 to 2.5 times more FAM-RIS than long-bone cultures, both after 4 and 24 h, and accordingly jaw osteoclasts were more susceptible to inhibition of prenylation of Rap1a after treatment with BPs for 24 h. Surprisingly, differences in BP uptake did not differentially affect osteoclastogenesis. This suggests that jaw osteoclast precursors are less sensitive to bisphosphonates after internalization. This was supported by the finding that gene expression of the anti-apoptotic genes Bcl-2 and Bcl-xL was higher in jaw cells than long bone cells, suggesting that the jaw cells might be more resistant to BP-induced apoptosis. Our findings suggest that bisphosphonates have distinct effects on both populations of osteoclast precursors and support previous findings that osteoclasts and precursors are bone-site specific. This study may help to provide more insights into bone-site-specific responses to bisphosphonates.
Co-reporter:Ernestas Gaidamauskas, Helen Parker, Boris A. Kashemirov, Alvin A. Holder, Kanokkarn Saejueng, Charles E. McKenna, Debbie C. Crans
Journal of Inorganic Biochemistry (December 2009) Volume 103(Issue 12) pp:1652-1657
Publication Date(Web):1 December 2009
DOI:10.1016/j.jinorgbio.2009.09.006
The coordination chemistry of bisphosphonates with Yb3+ was investigated to evaluate the potential of the UV–vis based detection method using the Yb3+–pyrocatechol complexation reaction as a sensor for bisphosphonates. The complexation chemistry of Yb3+ with phosphate and ATP analogs was previously described (E. Gaidamauskas, K. Saejueng, A.A. Holder, S. Bharuah, B.A. Kashemirov, D.C. Crans, C.E. McKenna, J. Biol. Inorg. Chem. 13 (2008) 1291–1299), and we here studied the complexation chemistry of bisphosphonates in this system. The spectrophotometric assay yields direct evidence for formation of a 4:3 metal to ligand complex at neutral pH. Direct evidence for Yb3+:methylenebis(phosphonate) complexes with 1:1 and 1:2 stoichiometry was also obtained by potentiometry at acidic and basic pH. Direct evidence for complex formation was obtained using 1H NMR spectroscopy although the stoichiometry was not accessed at neutral pH. Our results suggest that the spectroscopic observation of the YbPV complex can be used to conveniently measure concentrations of bisphosphonates down to 2–3 μM.
Co-reporter:Elena Ferri, Carlo Petosa, Charles E. McKenna
Biochemical Pharmacology (15 April 2016) Volume 106() pp:1-18
Publication Date(Web):15 April 2016
DOI:10.1016/j.bcp.2015.12.005
Co-reporter:Katarzyna M. Błażewska, Ralf Haiges, Boris A. Kashemirov, Frank H. Ebetino and Charles E. McKenna
Chemical Communications 2011 - vol. 47(Issue 22) pp:NaN6397-6397
Publication Date(Web):2011/05/09
DOI:10.1039/C1CC10876J
Under certain conditions, the phosphonocarboxylate analogue (3) of the bisphosphonate drug minodronate (4) in contact with borosilicate glassware reversibly forms an isolable dimer complex of boron, as revealed by the X-ray crystallographic structure of the (R,R/S,S) complex and supported by NMR and HRMS data.
.jpg)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4.png)
![(S)-tert-Butyl 2-(4-(4-chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepin-6-yl)acetate](http://img.cochemist.com/ccimg/1268600/1268524-70-4_b.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2.png)
![2-[(4S)-6-(4-CHLOROPHENYL)-8-METHOXY-1-METHYL-4H-[1,2,4]TRIAZOLO[4,3-A][1,4]BENZODIAZEPIN-4-YL]-N-ETHYLACETAMIDE](http://img.cochemist.com/ccimg/1261000/1260907-17-2_b.png)
![Spiro[isobenzofuran-1(3H),9'-[1H,5H,9H,10H,11H]xantheno[2,3,4-ij:5,6,7-i'j']diquinolizine]-ar-carboxylicacid, 2',3',6',7',12',13',16',17'-octahydro-3-oxo-, 2,5-dioxo-1-pyrrolidinylester](http://img.cochemist.com/ccimg/114700/114616-32-9.png)
![Spiro[isobenzofuran-1(3H),9'-[1H,5H,9H,10H,11H]xantheno[2,3,4-ij:5,6,7-i'j']diquinolizine]-ar-carboxylicacid, 2',3',6',7',12',13',16',17'-octahydro-3-oxo-, 2,5-dioxo-1-pyrrolidinylester](http://img.cochemist.com/ccimg/114700/114616-32-9_b.png)














![5'-Uridylic acid, anhydride with P,P'-methylenebis[phosphonic acid] (1:1)](/data/chemimg/1946300/71850-06-1.png)
![5'-Uridylic acid, anhydride with P,P'-methylenebis[phosphonic acid] (1:1)](/data/chemimg/1946300/71850-06-1_b.png)
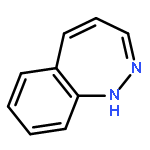
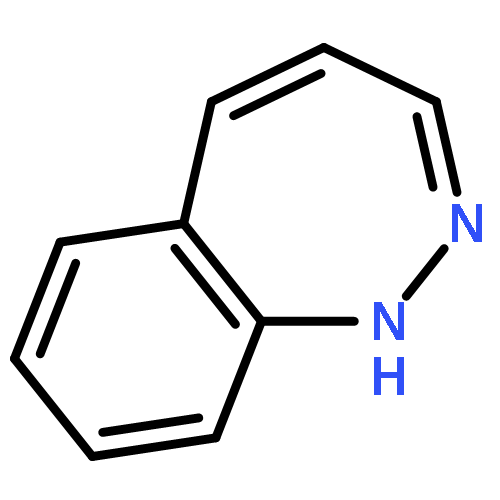


![L-Serine, L-valyl-O-[(5S)-5-[(4-amino-2-oxo-1(2H)-pyrimidinyl)methyl]-2-oxido-1,4,2-dioxaphosphorinan-2-yl]-, methyl ester](/data/chemimg/2002900/856686-24-3.png)
![L-Serine, L-valyl-O-[(5S)-5-[(4-amino-2-oxo-1(2H)-pyrimidinyl)methyl]-2-oxido-1,4,2-dioxaphosphorinan-2-yl]-, methyl ester](/data/chemimg/2002900/856686-24-3_b.png)


![2-Propenoic acid, 2-azido-3-imidazo[1,2-a]pyridin-3-yl-, ethyl ester, (Z)-](http://img.cochemist.com/ccimg/188400/188395-44-0.png)
![2-Propenoic acid, 2-azido-3-imidazo[1,2-a]pyridin-3-yl-, ethyl ester, (Z)-](http://img.cochemist.com/ccimg/188400/188395-44-0_b.png)
![(1-Hydroxy-2-(imidazo[1,2-a]pyridin-3-yl)ethane-1,1-diyl)diphosphonic acid](http://img.cochemist.com/ccimg/180100/180064-38-4.png)
![(1-Hydroxy-2-(imidazo[1,2-a]pyridin-3-yl)ethane-1,1-diyl)diphosphonic acid](http://img.cochemist.com/ccimg/180100/180064-38-4_b.png)
![Phosphonic acid,P-[[(1R)-2-(2,6-diamino-9H-purin-9-yl)-1-methylethoxy]methyl]-](http://img.cochemist.com/ccimg/147100/147057-10-1.png)
![Phosphonic acid,P-[[(1R)-2-(2,6-diamino-9H-purin-9-yl)-1-methylethoxy]methyl]-](http://img.cochemist.com/ccimg/147100/147057-10-1_b.png)


![L-Tyrosine, N-[(1,1-dimethylethoxy)carbonyl]-, 1-methylethyl ester](http://img.cochemist.com/ccimg/126600/126530-25-4.png)
![L-Tyrosine, N-[(1,1-dimethylethoxy)carbonyl]-, 1-methylethyl ester](http://img.cochemist.com/ccimg/126600/126530-25-4_b.png)


![9H-Purin-6-amine,9-[[(5S)-2-hydroxy-2-oxido-1,4,2-dioxaphosphorinan-5-yl]methyl]-](http://img.cochemist.com/ccimg/113900/113892-17-4.png)
![9H-Purin-6-amine,9-[[(5S)-2-hydroxy-2-oxido-1,4,2-dioxaphosphorinan-5-yl]methyl]-](http://img.cochemist.com/ccimg/113900/113892-17-4_b.png)
![3-(Chloromethyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/113900/113855-44-0.png)
![3-(Chloromethyl)imidazo[1,2-a]pyridine](http://img.cochemist.com/ccimg/113900/113855-44-0_b.png)






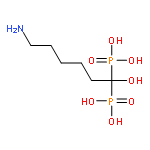





![L-Serine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl]-, methyl ester](http://img.cochemist.com/ccimg/71100/71017-97-5.png)
![L-Serine, N-[N-[(1,1-dimethylethoxy)carbonyl]-L-alanyl]-, methyl ester](http://img.cochemist.com/ccimg/71100/71017-97-5_b.png)
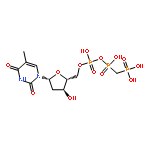
















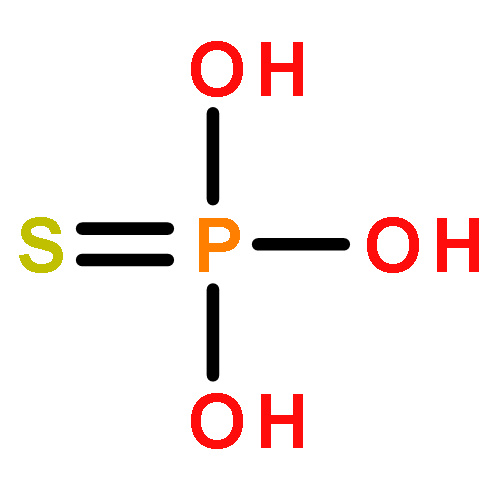






![2-[[DICHLORO-DI(PROPAN-2-YLOXY)PHOSPHORYLMETHYL]-PROPAN-2-YLOXYPHOSPHORYL]OXYPROPANE](http://img.cochemist.com/ccimg/10600/10596-22-2.png)
![2-[[DICHLORO-DI(PROPAN-2-YLOXY)PHOSPHORYLMETHYL]-PROPAN-2-YLOXYPHOSPHORYL]OXYPROPANE](http://img.cochemist.com/ccimg/10600/10596-22-2_b.png)








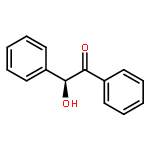
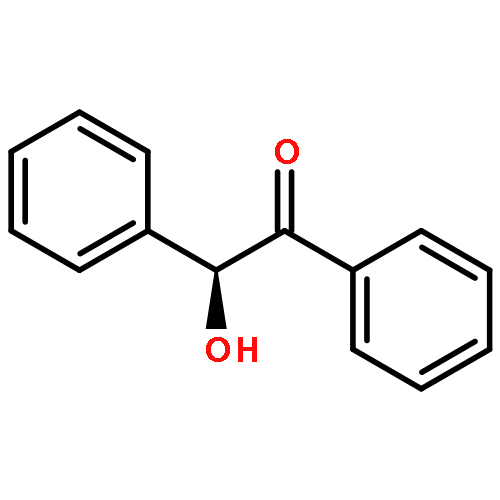


![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)
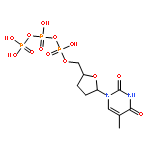



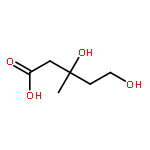













![SERINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-L-VALYL-, METHYL ESTER](http://img.cochemist.com/ccimg/71100/71017-98-6.png)
![SERINE, N-[(1,1-DIMETHYLETHOXY)CARBONYL]-L-VALYL-, METHYL ESTER](http://img.cochemist.com/ccimg/71100/71017-98-6_b.png)










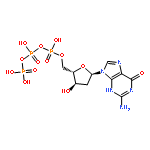




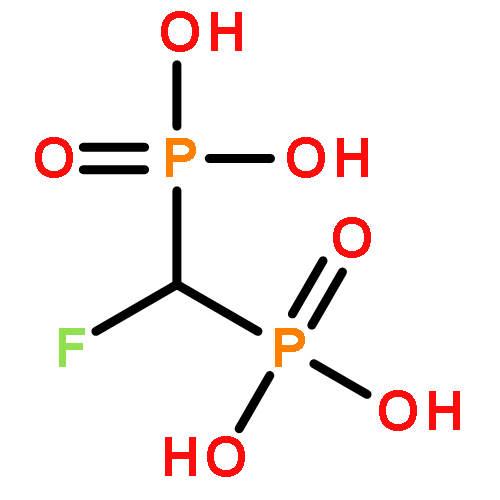
![Phosphonic acid,P-[[(1S)-2-(6-amino-9H-purin-9-yl)-1-(hydroxymethyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/93000/92999-29-6.png)
![Phosphonic acid,P-[[(1S)-2-(6-amino-9H-purin-9-yl)-1-(hydroxymethyl)ethoxy]methyl]-](http://img.cochemist.com/ccimg/93000/92999-29-6_b.png)