Co-reporter:Meixiang Gao;Minhua Zhang;Yonghui Li
RSC Advances (2011-Present) 2017 vol. 7(Issue 20) pp:11929-11937
Publication Date(Web):2017/02/16
DOI:10.1039/C6RA27610E
Mg–Al catalysts were adopted in the direct synthesis of 1,3-butadiene and other bulk chemicals from bioethanol. The influence of MgO content and sample calcination temperature on the catalytic performance was investigated. The optimization of reaction conditions for 1,3-butadiene, ethylene and 1-butanol, including reaction temperatures and the feed rate of ethanol, was also carried out. The catalysts were characterized by X-ray diffraction, nitrogen sorption, SEM, NH3-TPD, CO2-TPD and FTIR of adsorbed pyridine. On the basis of these analysis results and catalytic performance of yielding 1,3-butadiene, both the MgO content and the calcination temperature play a great role on the nature of acid and basic sites, then the proper active sites for producing 1,3-butadiene and other important chemicals such as ethylene and 1-butanol have been elaborated. The balance of moderate acid–basic sites is crucial to production of 1,3-butadiene from ethanol. Ethylene is obtained on strong acid and strong basic sites. 1-Butanol tends to form on the strong basic sites. To further improve the target product selectivity, modifiers adjusting the balance of acidity–basicity need to be involved in the Mg–Al catalysts.
Co-reporter:Fan HeJianxiong Wang, Yonghong Li, Hongwei Sun
Industrial & Engineering Chemistry Research 2017 Volume 56(Issue 7) pp:
Publication Date(Web):January 24, 2017
DOI:10.1021/acs.iecr.7b00307
Computational studies at the M06/6-311G(d,p) and M06-2X/6-311+G(d,p) levels were performed to explore the detailed mechanism of isoquinoline ring-opening and denitrogenation in a supercritical water system. Three reaction paths with the same product, 2-(2-oxoethyl) benzaldehyde, were supported by the computational results. The rate-limiting step in the major degradation reaction is an addition reaction at the N position. H2O is added to both the 1C–2N double bond (1C–2N addition reaction) and the 2N–3C double bond (2N–3C addition reaction) of the isoquinoline molecule, where the oxygen of H2O is added to the carbon atom. The energy barrier of the 1C–2N addition reaction is 52.7 kcal/mol, while that of 2N–3C addition (from Path 6) is 60.1 kcal/mol. From catalysis by two water molecules, the barrier of 1C–2N addition (Reaction (1)) is reduced to 27.5 kcal/mol. Catalysis from water molecule clusters is shown to considerably affect the process of isoquinoline ring-opening and denitrogenation, as indicated by comparing the reaction energy barrier heights with and without water catalysts.
Co-reporter:You Li, Xingang Li, Wentao Zhou, Hong Li, ... Yonghong Li
Journal of the Taiwan Institute of Chemical Engineers 2017 Volume 70(Volume 70) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.jtice.2016.10.048
•Experiments are performed and a kinetic controlled model is proposed.•Simultaneous method is employed for parameter regression.•Temperature evolution of emulsion is considered.•Partial water transformation is observed.•At high pressure, agitation should be enhanced for rapid hydrate formation.The hydrate formation kinetics of ethylene in water-in-oil (w/o) emulsion were measured with the pressure−volume−temperature (PVT) method during isochoric processes in an agitated reactor. Effects of operating conditions on ethylene hydrate formation are investigated systematically, including bath temperature, pressure, water content and standard volume ratio. A kinetic controlled model is proposed which employs the difference between equilibrium temperature and system temperature as the driving force. The kinetic parameters are regressed using some of the experimental data. Then the kinetic model is used to predict rest of the experimental results. The predictions and experimental data have shown good agreement. Nevertheless, there are two issues that deserve attention. First, it is found water is not completely transformed into hydrate, even though the temperature is below equilibrium. Second, when pressure is raised, for example over 15 bar, the pressure curve begins to drop slower than expectation. Acceleration in impeller agitation could alleviate the slowdown. The phenomenon is explained by the control step switch between inter-phase mass transfer and intrinsic kinetics.Download high-res image (139KB)Download full-size image
Co-reporter:Ying Ha;Benshuai Guo
Journal of Chemical Technology and Biotechnology 2016 Volume 91( Issue 2) pp:490-506
Publication Date(Web):
DOI:10.1002/jctb.4604
ABSTRACT
BACKGROUND
The issues of gasoline desulfurization are becoming more important because the regulated sulfur limits are being set lower and lower. Among the new desulfurization technologies, the olefin alkylation of thiophenic sulfur (OATS) process is more attractive than hydrodesulfurization because of its mild operating conditions and low loss of octane number. In this work, a sensitivity analysis and an economic analysis of a catalytic distillation process for alkylation desulfurization of fluid catalytic cracking (FCC) gasoline are presented. The objective is to evaluate the effects of various design and operating parameters on the OATS process by using a realistic FCC gasoline fraction.
RESULTS
A sensitivity analysis performed for operating pressure, reflux ratio, bottom flow rate, feed conditions, number of separate stages, number of reactive stages, catalyst weight, feed location, reactive zone position, and an economic analysis based on total annual costs (TAC) for these variables were studied.
CONCLUSION
The results show the qualitative environmental and economic effect of several design variables and operating conditions on the OATS process and provide a direction to the further design. In addition, more attention should be paid to feed flow rate control. © 2014 Society of Chemical Industry
Co-reporter:Xue Wang, Weiwei Xu, Yonghong Li, Jianxiong Wang, Fan He
Fluid Phase Equilibria 2016 Volume 409() pp:30-36
Publication Date(Web):15 February 2016
DOI:10.1016/j.fluid.2015.09.030
•Phase equilibria on three binary systems containing 2,5-dimethylthiophene and 2-ethylthiophene in hydrocarbons was measured with a circulating still.•Verify the consistency of the experimental data.•The binary systems exhibited positive deviation from ideal system and no azeotropic behavior was found.•The VLE data were correlated well by the activity coefficient models.Isobaric vapor–liquid equilibrium (VLE) data for binary systems 2-ethylthiophene + n-decane, 2,5-dimethylthiophene + n-decane and 2-ethylthiophene + mesitylene were measured at 101.33 kPa using the modified Rose–Williams equilibrium still. Gas chromatography was used to analyze compositions of the samples from the vapor–liquid equilibrium system. Thermodynamic consistency of the experimental data were checked both with the Herington consistency test and point-to-point test of Van Ness, which ensure the reliability of experimental data. The experimental data were correlated by the Wilson and non-random two-liquid (NRTL) activity coefficient models and were compared with original UNIFAC predictive model. The corresponding parameters for the Wilson and NRTL models were obtained. The results show that the calculated values of the vapor-phase mole fraction and bubble point temperature by Wilson, NRTL and UNIFAC models agree well with the experimental data.
Co-reporter:Xiaojiao Liu, Yonghong Li and Ranran Zhang
RSC Advances 2015 vol. 5(Issue 104) pp:85453-85459
Publication Date(Web):24 Sep 2015
DOI:10.1039/C5RA16072C
A series of CHA-type trimetallic composite zeolites of Cex–Fey/Cu–SSZ-13 catalysts were prepared using Cu–SSZ-13, synthesized in a one-pot procedure, and subsequent ion-exchange with Fe3+ and Ce3+. The catalysts were characterized including TEM, XRD, XPS, SEM and BET. Their catalytic performances for selective catalytic reduction of NO with NH3 were investigated. The results of XRD revealed that the crystal structure of zeolite Ce–Fe/Cu–SSZ-13 is the same as Cu–SSZ-13. It is known from the results of BET and catalytic performance tests that larger specific surface areas and smaller pore size favor a catalytic reaction. Among the prepared Cex–Fey/Cu–SSZ-13 catalysts, Ce0.017–Fe0.017/Cu–SSZ-13 displayed the best SCR performance. The NO conversion was more than 90% between 200 and 500 °C. N2 selectivity was above 98% within the wider temperature range of 150–550 °C. In addition, the catalyst demonstrated sulfur–water tolerance and effective resistance against high space velocity. The phenomena suggest that synergistic effects of Cu, Fe and Ce species improve the SCR performances and make the Ce–Fe/Cu–SSZ-13 catalyst a promising candidate for NH3–SCR technology.
Co-reporter:Ying Ha
Journal of Porous Materials 2015 Volume 22( Issue 3) pp:721-728
Publication Date(Web):2015 June
DOI:10.1007/s10934-015-9945-5
The alkylation of 2-ethylthiophene with vinyltoluene in the model crack C9 fraction over tungstophosphoric acid supported on four solids (SiO2, nanoSiO2, TiO2 and nanoTiO2) was investigated. It was found that tungstophosphoric acid supported on nanoSiO2 exhibited excellent catalytic performance in this desulfurization process. These catalysts were characterized by FT-IR, XRD, BET, NH3-TPD and TGA. The results showed that both the acidity and the textural properties of the catalysts had significant influences on the catalytic performance. Moreover, the influence of catalyst loading was also studied. Meanwhile, the reaction parameters, including the reaction temperature, the catalyst dosage and reaction time, were optimized.
Co-reporter:Xue Wang, Yonghong Li
Fluid Phase Equilibria 2014 Volume 378() pp:113-117
Publication Date(Web):25 September 2014
DOI:10.1016/j.fluid.2014.07.010
•Isobaric vapor–liquid equilibrium for binary system of 2-ethylthiophene + n-octane was measured with a circular still.•The binary system exhibited positive deviation from ideal system and no azeotropic behavior was found.•The experimental data were correlated with the Wilson, NRTL and UNIQUAC models.•All VLE measurement passed the thermodynamic consistency test.Isobaric vapor–liquid equilibrium (VLE) data of 2-ethylthiophene and n-octane binary system were measured at 101.33 kPa with a modified Rose-Williams still. Gas chromatography was used to analyze compositions of the samples from the vapor–liquid equilibrium system. The VLE measurement passed the thermodynamic consistency test proposed by Herington and no azeotropic behavior was found. The experimental data were correlated by Wilson, the non-random two-liquid (NRTL) and universal quasi-chemical activity coefficient (UNIQUAC) models, respectively. The corresponding parameters for the three models were obtained. Results showed that the Wilson model gave better predictions than NRTL and UNIQUAC models.
Co-reporter:Xiaoliang Li, Yonghong Li
Journal of Molecular Catalysis A: Chemical 2014 Volume 386() pp:69-77
Publication Date(Web):May 2014
DOI:10.1016/j.molcata.2014.02.016
•The MoO3 modified CeAlOx catalyst exhibited excellent NH3-SCR catalytic activity and high resistance to SO2 or/and H2O poisoning at 250 °C.•The redox capacity and total acidity of the catalyst were enhanced by the introduction of Mo.•A possible reaction mechanism was proposed over the CeMo0.5AlOx catalyst.A series of MoO3 doped CeAlOx mixed oxide catalysts with different Mo/Al molar ratios were prepared by the simple coprecipitation method and used for selective catalytic reduction of NO with NH3. The Ce-Mo-AlOx catalyst with the Mo/Al molar ratio of 0.5 exhibited excellent activity and high H2O or/and SO2 poisoning resistance at 250 °C. XRD revealed that the molybdenum oxide existed in either highly dispersed or amorphous phases on the catalyst surface. BET analysis results showed that the total pore volume and the average pore diameter of the CeAlOx catalyst was improved by the addition of MoO3. As determined by the H2-TPR and NH3-TPD, the redox capacity and total acidity of the CeAlOx catalyst were also enhanced by the introduction of MoO3, which are critical for the NH3-SCR reaction. The SCR reaction mechanism was also studied by the in situ DRIFTS, the coordinated NH3 and ionic NH4+ species together with the monodentate and bidentate nitrate were active intermediates on the CeMo0.5AlOx catalyst surface during the NH3-SCR reaction.MoO3 modified CeAlOx catalyst prepared by the facile coprecipitation method has been utilized for the selective catalytic reduction of NO with NH3. The catalyst showed excellent activity, prominent resistant to the space velocity and exhibited high SO2 or/and H2O durability at 250 °C.
Co-reporter:Xiaoliang Li
Reaction Kinetics, Mechanisms and Catalysis 2014 Volume 112( Issue 1) pp:27-36
Publication Date(Web):2014 June
DOI:10.1007/s11144-014-0688-0
CeO2–MoO3 catalysts were prepared by three methods: single step sol–gel, homogeneous precipitation, hydrothermal. These catalysts were used for the selective catalytic reduction of NO with NH3 in the temperature range from 150 to 400 °C. The tested results suggested that the catalyst prepared by the hydrothermal method showed the higher NO conversion. The BET, XRD, H2-TPR, NH3-TPD and in situ DRIFTS characterization results indicated that the larger surface area, highly dispersed nanoctrystalline ceria, good redox ability and the stronger adsorption capacity for the NH3 should be the key factors to acquire the better NH3-SCR performance.
Co-reporter:Benshuai Guo, Juan Bai, Yonghong Li, Shuqian Xia, Peisheng Ma
Fluid Phase Equilibria 2013 Volume 353() pp:87-92
Publication Date(Web):15 September 2013
DOI:10.1016/j.fluid.2013.05.019
•Isobaric VLE data for four systems was measured in dilute solution.•The VLE data in dilute solution were compared with that in the entire concentration.•The activity coefficients of trace component varied a lot in its low concentration.•The y–x of the trace component followed the quadratic polynomial equation.•A concept of multi-scale phase equilibrium was proposed.Isobaric vapor–liquid equilibrium (VLE) data for the binary systems of thiophenic compounds with three hydrocarbons in gasoline (2,3-dimethyl-1-butene, 2,3-dimethyl-2-butene and n-heptane) were measured in dilute solutions at 101.33 kPa with a modified Rose–Williams still. General Coulomb meter was used to analyze the compositions of the samples from the vapor–liquid equilibrium systems. The VLE data was compared with the calculated value by the Wilson model with the interaction parameters regressed over the entire concentration range. Results found that neither the activity coefficient model nor the Henry's model could describe the VLE behavior of the trace component in the dilute solution accurately. Thus a concept of multi-scale phase equilibrium was proposed.
Co-reporter:Aihua Kong;Yanyu Wei
Frontiers of Chemical Science and Engineering 2013 Volume 7( Issue 2) pp:170-176
Publication Date(Web):2013 June
DOI:10.1007/s11705-013-1322-9
A high-performance Ni/ZnO adsorbent was prepared by homogeneous precipitation using urea hydrolysis and characterized by N2 adsorption-desorption, X-ray diffraction (XRD), and scanning electron microscope (SEM). The adsorbent was applied to the deep desulfurization of gasoline and showed a high breakthrough sulfur capacity and a remarkably high volume hourly space velocity. The effects of coexisting olefins in gasoline as well as adsorptive conditions on the adsorptive performance were examined. It was found that olefins in gasoline had a slightly inhibiting effect on the desulfurization performance of the adsorbent. The optimum conditions were 673 K, 1.0 Mpa with a volume hourly space velocity of 60 h−1. Under the optimum conditions, ultralow sulfur gasoline could be produced and the breakthrough sulfur capacity of the adsorbent was 360 mg-s/g-sorb for the model gasoline.
Co-reporter:Rong Wang, Yonghong Li, Benshuai Guo, and Hongwei Sun
Energy & Fuels 2011 Volume 25(Issue 9) pp:3940
Publication Date(Web):August 12, 2011
DOI:10.1021/ef200705b
The desulfurization of fluid catalytic cracking (FCC) gasoline by alkylation over solid acid catalysts is considered to be a viable and less costly path to meet environmental regulations of sulfur emissions. However, side reactions in the process lead to significant levels of coke, which will greatly reduce the lifetime of the catalyst. In this paper, the catalytic mechanism of MCM-41 supported phosphoric acid catalyst for gasoline desulfurization by alkylation has been investigated by using experimental methods and quantum chemical calculations to study the catalytic behavior for the adsorption and reaction of different reactants, which can help optimize the reaction conditions and preparation methods of the catalyst for a more efficient alkylation process. The results showed that both the typical main and side reactions in the alkylation process started from a stable alkoxide intermediate that was formed by protonation of olefin adsorbed on the catalyst. Thiophenic compounds were more inclined to be adsorbed on the alkoxide intermediate than olefins for further reaction, and the activation energy for the alkylation of thiophenic sulfurs with alkenes was obviously lower than that for alkene oligomerization. Moreover, the thiophene alkylation was exothermic while the olefin oligomerization was endothermic. On the basis of these findings obtained by experimental and theoretical investigation, two methods that might be useful to further inhibit the occurrence of side reactions and improve the catalyst performance in the alkylation process were proposed.
Co-reporter:Chuanzhuang Feng, Jilian Dong, Yonghong Li
Fluid Phase Equilibria 2011 Volume 309(Issue 2) pp:201-205
Publication Date(Web):25 October 2011
DOI:10.1016/j.fluid.2011.07.014
Isothermal vapor–liquid equilibrium (VLE) data were measured for the binary system methanol and 2,3-dimethyl-2-butene at 343.15 K, 353.15 K, 363.15 K, 373.15 K, respectively. The measurements were carried out in a novel recirculation equilibrium equipment. Three activity coefficient models including Wilson, NRTL and UNIQUAC, as well as the Soave–Redlich–Kwong equation of state were used to correlate the experimental data. The correlation results showed that a good consistency between the experimental data and the Wilson model can be achieved.Highlights► Isothermal VLE data for methanol + 2,3-dimethyl-2-butene were measured at 343.15 K, 353.15 K, 363.15 K, 373.15 K. ► Parameters of Wilson, NRTL and UNIQUAC models at different temperatures were regressed. ► Wilson model gives the best correlation of the VLE data. ► The binary system shows a positive deviation from Raoult's law and has a maximum pressure azeotrope.
Co-reporter:Rong Wang, Yonghong Li, Ronghui Shi, Meimei Yang
Journal of Molecular Catalysis A: Chemical 2011 Volume 344(1–2) pp:122-127
Publication Date(Web):17 June 2011
DOI:10.1016/j.molcata.2011.05.009
The effect of the interaction between metal and support on the performance of different phases alumina supported nickel catalysts was demonstrated in liquid phase selective hydrogenation of isoprene in simulated gasoline. Catalysts supported on γ-Al2O3 and κ-Al2O3 were characterized by BET, XRD, TPR, and XPS techniques. The results showed that a “surface spinel”, NiAl2O4 was formed. The supported nickel ions preferentially incorporate into the tetrahedral vacancies of γ-Al2O3 support to form SMSI (strong metal–support interaction), while WMSI (weak metal–support interaction) was formed on κ-Al2O3 support with little tetrahedral vacancy. When there were enough hydrogenation sites, the Ni/γ-Al2O3 catalysts with SMSI which can resist carbon deposition, performed higher isoprene conversion, higher stability and lower selectivity than the same nickel loading Ni/κ-Al2O3 samples with WMSI. The weak interaction had a positive effect on the formation of coke, which was mainly related to the hydrogenolytic sites, leading to main reaction with high mono-olefins selectivity with isoprene conversion decrease evidently.Graphical abstractComparable studies of metal–support interaction with γ-Al2O3 and κ-Al2O3 phases effects on catalytic performance of Ni/Al2O3 for selective hydrogenation of isoprene have been carried out in simulated gasoline. The degree of the interaction connected with the nickel content and alumina phase affects coke deposition and hydrogenation sites deactivation significantly, leading to the difference of the selectivity of mono-olefins in selective hydrogenation.Highlights► The different metal–support interactions over two alumina supports were characterized. ► The effect of the different interaction on catalytic performance was explained. ► The weak interaction resisting coke deposition was related to the hydrogenolytic sites.
Co-reporter:Jilian Dong, Chuanzhuang Feng, and Yonghong Li
Journal of Chemical & Engineering Data 2011 Volume 56(Issue 5) pp:2386-2392
Publication Date(Web):March 10, 2011
DOI:10.1021/je101304x
Isothermal vapor−liquid equilibrium (VLE) data of the methanol and 2,3-dimethyl-1-butene binary system were measured at 343.06 K, 353.27 K, 363.19 K, and 372.90 K. These measurements were made with novel recirculation equilibrium stainless steel equipment. Gas chromatography was used to analyze compositions of the samples from the vapor−liquid equilibrium systems. The binary VLE data were correlated with various activity coefficient models including Wilson, NRTL, and UNIIQUAC, as well as the Soave−Redlich−Kwong equation of state.
Co-reporter:Benshuai Guo, Rong Wang, Yonghong Li
Fuel 2011 Volume 90(Issue 2) pp:713-718
Publication Date(Web):February 2011
DOI:10.1016/j.fuel.2010.10.010
Gasoline desulfurization is receiving attention worldwide due to the increasing stringent regulations on sulfur content for environmental protection purpose. As conventional hydrotreating technology leads to significant octane number loss and processing costs, the gasoline alkylation desulfurization process, which consists of weighing down the sulfuric compounds by catalytic alkylation with olefins present in the feed and distillation followed by, is a rather attractive way. In this paper, firstly alkylation of thiophenic compounds was researched over macroporous sulfonic resin Amberlyst 35 in methanol presence to increase the selectivity of catalyst, then kinetics of thiophenic sulfurs alkylation in FCC gasoline was researched without and with methanol. Results found that appropriate methanol (⩽2 wt.% methanol in model gasoline and ⩽1 wt.% methanol in FCC gasoline) could inhibit olefins oligomerization significantly without influence on the conversions of thiophenic compounds. The alkylation of thiophenic sulfurs could be described as pseudo first order reaction regardless of the existence of methanol. The introduction of methanol decreases the reaction rate constant and increases the activation energy of alkylation reactions.
Co-reporter:Benshuai Guo, Rong Wang, Yonghong Li
Fuel Processing Technology 2010 Volume 91(Issue 11) pp:1731-1735
Publication Date(Web):November 2010
DOI:10.1016/j.fuproc.2010.07.012
Sulfur removal has received increasing attention in recent years primarily for environmental protection purpose. As an attractive technology in the case of gasoline, OATS (olefinic alkylation of thiophenic sulfur) proposed to separate sulfur compounds by distillation after being weighed down by alkylation with olefins in the feed. In this paper, alkylation reactions of thiophenic compounds were studied over solid phosphoric acid catalysts (SPAM and SPAS using MCM-41 and Silicalite-1 zeolite as supporters respectively) and macroporous sulfonic resins (including NKC-9, D005-2 and Amberlyst 35) with model gasoline and FCC (fluid catalytic cracking) gasoline. Results showed that macroporous sulfonic resins showed better performance than solid phosphoric acid catalysts under milder conditions in both feeds. Among the resins, Amberlyst 35 was the most suitable catalyst for the application of catalytic distillation for its good performance at the temperature range of 353–413 K in FCC gasoline. However, the selectivity of isoamylene dimerization over Amberlyst 35 decreased with the temperature, which was harmful to the product yield and catalyst stability. Besides, different activity orders of solid phosphoric acid catalysts in model gasoline and FCC gasoline were explained by combining the acidic properties of the catalysts with the species of olefins in two feeds.
Co-reporter:Ronghui Shi;Rong Wang;Benshuai Guo
Catalysis Letters 2010 Volume 139( Issue 3-4) pp:114-122
Publication Date(Web):2010 November
DOI:10.1007/s10562-010-0412-2
The alkylation of thiophenic compounds with olefins in real fluid catalytic cracking (FCC) gasoline was carried out over three kinds of acidic catalysts: HY zeolite, phosphoric acid supported on both kieselguhr and purely siliceous MCM-41 zeolite. The results showed that the MCM-41 supported phosphoric acid catalyst was the most promising to be utilized in the reaction in terms of its activity and stability. Based on its excellent catalytic performance, apparent kinetic models for alkylation of the major thiophenic sulfides were proposed, thus allowing use of the simulation in reaction distillation (RD) technology. Furthermore, numerical regression led to kinetic parameters with narrow spans, suggesting that the proposed models satisfactorily simulate the sulfides removal under the suggested reaction conditions.

![2,5-Piperazinedione,3-hydroxy-3-[(3-hydroxyphenyl)methyl]-1,4-dimethyl-6-[(4-nitro-1H-indol-3-yl)methyl]-,(3R,6S)-](http://img.cochemist.com/ccimg/122400/122380-18-1.png)
![2,5-Piperazinedione,3-hydroxy-3-[(3-hydroxyphenyl)methyl]-1,4-dimethyl-6-[(4-nitro-1H-indol-3-yl)methyl]-,(3R,6S)-](http://img.cochemist.com/ccimg/122400/122380-18-1_b.png)


![Pyridazine, 3-chloro-6-methyl-4-[3-(trifluoromethyl)phenyl]-](/data/chemimg/103200/938189-00-5.png)
![Pyridazine, 3-chloro-6-methyl-4-[3-(trifluoromethyl)phenyl]-](/data/chemimg/103200/938189-00-5_b.png)






















![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-3-hydroxy-](http://img.cochemist.com/ccimg/90900/90819-30-0.png)
![L-Phenylalanine,N-[(1,1-dimethylethoxy)carbonyl]-3-hydroxy-](http://img.cochemist.com/ccimg/90900/90819-30-0_b.png)








![4-AMINO-5-PYRIDIN-3-YL-4H-[1,2,4]TRIAZOLE-3-THIOL](http://img.cochemist.com/ccimg/78100/78027-00-6.png)
![4-AMINO-5-PYRIDIN-3-YL-4H-[1,2,4]TRIAZOLE-3-THIOL](http://img.cochemist.com/ccimg/78100/78027-00-6_b.png)
![1,2-Benzisothiazol-3(2H)-one, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-,1,1-dioxide](http://img.cochemist.com/ccimg/75000/74921-64-5.png)
![1,2-Benzisothiazol-3(2H)-one, 2-[2-(4-methoxyphenyl)-2-oxoethyl]-,1,1-dioxide](http://img.cochemist.com/ccimg/75000/74921-64-5_b.png)

















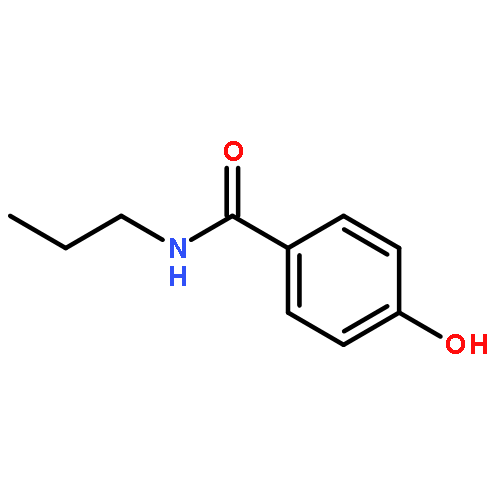
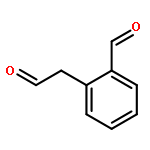
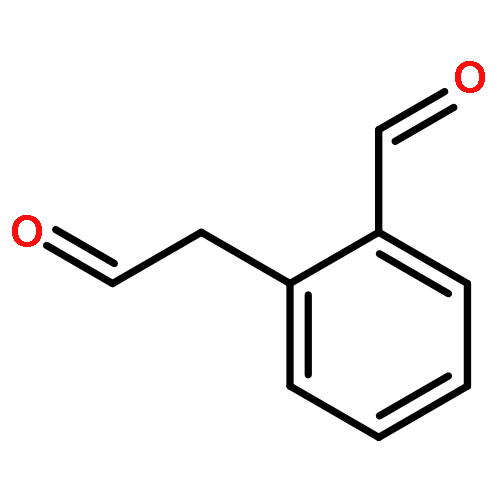
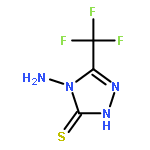











![2-(Hydroxymethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide](http://img.cochemist.com/ccimg/14000/13947-20-1.png)
![2-(Hydroxymethyl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide](http://img.cochemist.com/ccimg/14000/13947-20-1_b.png)
![ethyl [(4-chlorophenyl)sulfonyl]carbamate](http://img.cochemist.com/ccimg/14000/13945-53-4.png)
![ethyl [(4-chlorophenyl)sulfonyl]carbamate](http://img.cochemist.com/ccimg/14000/13945-53-4_b.png)















![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5.png)
![4H-PYRAZOLO[1,5-C][1,3]THIAZOLE-2-CARBALDEHYDE](http://img.cochemist.com/ccimg/3300/3296-03-5_b.png)








![[[3-[(Dimethylamino)carbonyl]-2-pyridinyl]sulfonyl]carbamic acid ethyl ester](http://img.cochemist.com/ccimg/144100/144098-17-9.png)
![[[3-[(Dimethylamino)carbonyl]-2-pyridinyl]sulfonyl]carbamic acid ethyl ester](http://img.cochemist.com/ccimg/144100/144098-17-9_b.png)
![Carbamic acid, [(2-chlorophenyl)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/90100/90096-45-0.png)
![Carbamic acid, [(2-chlorophenyl)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/90100/90096-45-0_b.png)


![Benzoic acid, 2-[[(ethoxycarbonyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/83500/83404-84-6.png)
![Benzoic acid, 2-[[(ethoxycarbonyl)amino]sulfonyl]-, methyl ester](http://img.cochemist.com/ccimg/83500/83404-84-6_b.png)




![Carbamic acid, [(2-nitrophenyl)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/22900/22819-25-6.png)
![Carbamic acid, [(2-nitrophenyl)sulfonyl]-, ethyl ester](http://img.cochemist.com/ccimg/22900/22819-25-6_b.png)







