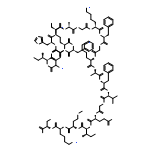
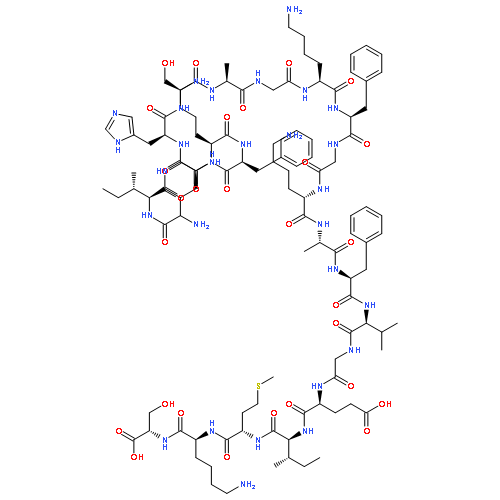
Co-reporter:Chuhan Zong, Michelle J. Wu, Jason Z. Qin, and A. James Link
Journal of the American Chemical Society August 2, 2017 Volume 139(Issue 30) pp:10403-10403
Publication Date(Web):July 11, 2017
DOI:10.1021/jacs.7b04830
Mechanically interlocked molecules that change their conformation in response to stimuli have been developed by synthetic chemists as building blocks for molecular machines. Here we describe a natural product, the lasso peptide benenodin-1, which exhibits conformational switching between two distinct threaded conformers upon actuation by heat. We have determined the structures of both conformers and have characterized the kinetics and energetics of the conformational switch. Single amino acid substitutions to benenodin-1 generate peptides that are biased to a single conformer, showing that the switching behavior is potentially an evolvable trait in these peptides. Lasso peptides such as benenodin-1 can be recognized and cleaved by enzymes called lasso peptide isopeptidases. We show that only the native conformer of benenodin-1 is cleaved by its cognate isopeptidase. Thus, thermally induced conformational switching of benenodin-1 may also be relevant to the biological function of these molecules.
Co-reporter:Jonathan R. Chekan, Joseph D. Koos, Chuhan Zong, Mikhail O. Maksimov, A. James LinkSatish K. Nair
Journal of the American Chemical Society 2016 Volume 138(Issue 50) pp:16452-16458
Publication Date(Web):November 27, 2016
DOI:10.1021/jacs.6b10389
Lasso peptides are a class of bioactive ribosomally synthesized and post-translationally modified peptides (RiPPs), with a threaded knot structure that is formed by an isopeptide bond attaching the N-terminus of the peptide to a side chain carboxylate. Some lasso peptide biosynthetic clusters harbor an enzyme that specifically hydrolyzes the isopeptide bond to yield the linear peptide. We describe here the 2.4 Å resolution structure of a lasso peptide isopeptidase revealing a topologically novel didomain architecture consisting of an open β-propeller appended to an α/β hydrolase domain. The 2.2 Å resolution cocrystal structure of an inactive variant in complex with a lasso peptide reveals deformation of the substrate, and reorganization of the enzyme active site, which exposes and orients the isopeptide bond for hydrolysis. Structure-based mutational analysis reveals how this enzyme recognizes the lasso peptide substrate by shape complementarity rather than through sequence specificity. The isopeptidase gene can be used to facilitate genome mining, as a network-based mining strategy queried with this sequence identified 87 putative lasso peptide biosynthetic clusters, 65 of which have not been previously described. Lastly, we validate this mining approach by heterologous expression of two clusters encoded within the genome of Asticcaucalis benevestitus, and demonstrate that both clusters produce lasso peptides.
Co-reporter:Caitlin D. Allen and A. James Link
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14214-14217
Publication Date(Web):October 19, 2016
DOI:10.1021/jacs.6b09454
Lasso peptides exist naturally in a threaded state as [1]rotaxanes, and we reasoned that lasso peptides cleaved in their loop region could serve as building blocks for catenanes. Mutagenesis of the lasso peptide microcin J25 (MccJ25) with two cysteine residues followed by cleavage of the peptide with trypsin led to a [2]rotaxane structure that self-assembled into a [3]catenane and [4]catenanes at room temperature in aqueous solution. The [3]catenane represents the smallest ring size of a catenane composed solely of polypeptide segments. The NMR structure of the [3]catenane was determined, suggesting that burial of hydrophobic residues may be a driving force for assembly of the catenane structure.
Co-reporter:Chuhan Zong, Mikhail O. Maksimov, and A. James Link
ACS Chemical Biology 2016 Volume 11(Issue 1) pp:61
Publication Date(Web):October 22, 2015
DOI:10.1021/acschembio.5b00745
Lasso peptides are a family of ribosomally synthesized and post-translationally modified peptides (RiPPs) typified by an isopeptide-bonded macrocycle between the peptide N-terminus and an aspartate or glutamate side chain. The C-terminal portion of the peptide threads through the N-terminal macrocycle to give the characteristic lasso fold. Because of the inherent stability, both proteolytic and often thermal, of lasso peptides, we became interested in whether proteins could be fused to the free C-terminus of lasso peptides. Here, we demonstrate fusion of two model proteins, the artificial leucine zipper A1 and the superfolder variant of GFP, to the C-terminus of the lasso peptide astexin-1. Successful lasso cyclization of the N-terminus of these fusion proteins requires a flexible linker in between the C-terminus of the lasso peptide and the N-terminus of the protein of interest. The ability to fuse lasso peptides to a protein of interest is an important step toward phage and bacterial display systems for the high-throughput screening of lasso peptide libraries for new functions.
Co-reporter:Caitlin D. Allen, Maria Y. Chen, Alexander Y. Trick, Dan Thanh Le, Andrew L. Ferguson, and A. James Link
ACS Chemical Biology 2016 Volume 11(Issue 11) pp:3043
Publication Date(Web):September 2, 2016
DOI:10.1021/acschembio.6b00588
Lasso peptides are a class of knot-like polypeptides in which the C-terminal tail of the peptide threads through a ring formed by an isopeptide bond between the N-terminal amine group and a side chain carboxylic acid. The small size (∼20 amino acids) and simple topology of lasso peptides make them a good model system for studying the unthreading of entangled polypeptides, both with experiments and atomistic simulation. Here, we present an in-depth study of the thermal unthreading behavior of two lasso peptides astexin-2 and astexin-3. Quantitative kinetics and energetics of the unthreading process were determined for variants of these peptides using a series of chromatography and mass spectrometry experiments and biased molecular dynamics (MD) simulations. In addition, we show that the Tyr15Phe variant of astexin-3 unthreads via an unprecedented “tail pulling” mechanism. MD simulations on a model ring-thread system coupled with machine learning approaches also led to the discovery of physicochemical descriptors most important for peptide unthreading.
Co-reporter:Wai Ling Cheung, Maria Y. Chen, Mikhail O. Maksimov, and A. James Link
ACS Central Science 2016 Volume 2(Issue 10) pp:702
Publication Date(Web):September 29, 2016
DOI:10.1021/acscentsci.6b00184
Lasso peptides are a member of the superclass of ribosomally synthesized and posttranslationally modified peptides (RiPPs). Like all RiPPs, lasso peptides are derived from a gene-encoded precursor protein. The biosynthesis of lasso peptides requires two enzymatic activities: proteolytic cleavage between the leader peptide and the core peptide in the precursor protein, accomplished by the B enzymes, and ATP-dependent isopeptide bond formation, accomplished by the C enzymes. In a subset of lasso peptide biosynthetic gene clusters from Gram-positive organisms, the B enzyme is split between two proteins. One such gene cluster is found in the organism Rhodococcus jostii, which produces the antimicrobial lasso peptide lariatin. The B enzyme in R. jostii is split between two open reading frames, larB1 and larB2, both of which are required for lariatin biosynthesis. While the cysteine catalytic triad is found within the LarB2 protein, LarB1 is a PqqD homologue expected to bind to the lariatin precursor LarA based on its structural homology to other RiPP leader peptide binding domains. We show that LarB1 binds to the leader peptide of the lariatin precursor protein LarA with a sub-micromolar affinity. We used photocrosslinking with the noncanonical amino acid p-azidophenylalanine and mass spectrometry to map the interaction of LarA and LarB1. This analysis shows that the LarA leader peptide interacts with a conserved motif within LarB1 and, unexpectedly, the core peptide of LarA also binds to LarB1 in several positions. A Rosetta model built from distance restraints from the photocrosslinking experiments shows that the scissile bond between the leader peptide and core peptide in LarA is in a solvent-exposed loop.
Co-reporter:Frank J. Piscotta, Jeffery M. Tharp, Wenshe R. Liu and A. James Link
Chemical Communications 2015 vol. 51(Issue 2) pp:409-412
Publication Date(Web):04 Nov 2014
DOI:10.1039/C4CC07778D
Using the amber suppression approach, four noncanonical amino acids (ncAAs) were used to replace existing amino acids at four positions in lasso peptide microcin J25 (MccJ25). The lasso peptide biosynthesis enzymes tolerated all four ncAAs and produced antibiotics with efficacy equivalent to wild-type in some cases. Given the rapid expansion of the genetically encoded ncAA pool, this study is the first to demonstrate an expedient method to significantly increase the chemical diversity of lasso peptides.
Co-reporter:Saw Kyin;Stanislav Y. Shvartsman;Alan S. Futran
PNAS 2015 Volume 112 (Issue 28 ) pp:8590-8595
Publication Date(Web):2015-07-14
DOI:10.1073/pnas.1501373112
Extracellular signal-regulated kinase (ERK) coordinates cellular responses to a range of stimuli by phosphorylating its numerous
substrates. One of these substrates, Capicua (Cic), is a transcriptional repressor that was first identified in Drosophila and has been implicated in a number of human diseases. Here we use a chemical biology approach to map the binding interface
of ERK and Cic. The noncanonical amino acid p-azidophenylalanine (AzF) was introduced into the ERK-binding region of Drosophila Cic, and photocrosslinking and tandem mass spectrometry were used to pinpoint its binding site on ERK. We also identified
the ERK-binding region of human Cic and showed that it binds to the same site on ERK despite lacking conservation with the
Drosophila Cic binding region. Finally, we mapped the amino acids involved in human Cic binding to ERK using AzF-labeled ERK. These
results reveal the molecular details of the ERK–Cic interaction and demonstrate that the photocrosslinking approach is complementary
to existing methods for mapping kinase–substrate binding interfaces.
Co-reporter:Diya M. Abdeljabbar, Frank J. Piscotta, Siyan Zhang and A. James Link
Chemical Communications 2014 vol. 50(Issue 94) pp:14900-14903
Publication Date(Web):09 Oct 2014
DOI:10.1039/C4CC06528J
Here we demonstrate a methodology, termed protein stapling, for the introduction of covalent constraints into recombinant proteins. Using the azide–alkyne click reaction as the stapling chemistry, we have improved the thermostability of a model leucine zipper protein. Additionally, stapling the core of the small, globular protein G resulted in improved binding to its target, immunoglobulin G.
Co-reporter:Mikhail O. Maksimov
Journal of Industrial Microbiology & Biotechnology 2014 Volume 41( Issue 2) pp:333-344
Publication Date(Web):2014 February
DOI:10.1007/s10295-013-1357-4
Genome mining has unlocked a veritable treasure chest of natural compounds. However, each family of natural products requires a genome-mining approach tailored to its unique features to be successful. Lasso peptides are ribosomally synthesized and posttranslationally modified products with a unique three-dimensional structure. Advances in the understanding of these molecules have informed the design of strategies to identify new members of the class in sequenced genomes. This review presents the bioinformatic methods used to discover novel lasso peptides and describes how such analyses have afforded insights into the biosynthesis and evolution of this peptide class.
Co-reporter:Mikhail O. Maksimov and A. James Link
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:12038-12047
Publication Date(Web):July 17, 2013
DOI:10.1021/ja4054256
Lasso peptides are a class of ribosomally derived natural products with diverse bioactivities. The characteristic threaded lasso structure in these peptides derives from an isopeptide bond attaching the N-terminus of the peptide to an acidic side chain. Here we describe the heterologous expression of a lasso peptide gene cluster encoding two lasso peptides, astexin-2 and astexin-3, and solve the solution structure of astexin-3. This cluster also encodes an enzyme annotated as a protease. We show that this enzyme, AtxE2, is a lasso peptide isopeptidase that specifically hydrolyzes astexins-2 and -3, converting them to linear peptides. Astexin-3 is highly thermostable and resists unthreading after extensive heat treatment. In contrast, astexin-2 unthreads upon heat treatment. AtxE2 has no activity toward unthreaded astexin-2, demonstrating that this isopeptidase must recognize a knotted structure in order to function. We also use this isopeptidase as a tool to study evolutionary relationships between lasso peptide gene clusters.
Co-reporter:Mikhail O. Maksimov, Si Jia Pan and A. James Link
Natural Product Reports 2012 vol. 29(Issue 9) pp:996-1006
Publication Date(Web):25 Jul 2012
DOI:10.1039/C2NP20070H
Covering: up to May 2012
Lasso peptides are a class of ribosomally-synthesized and posttranslationally-modified natural products with diverse bioactivities. This review describes the structure and function of all known lasso peptides (as of mid-2012) and covers our current knowledge about the biosynthesis of those molecules. The isolation and characterization of lasso peptides are also covered as are bioinformatics strategies for the discovery of new lasso peptides from genomic sequence data. Several studies on the engineering of new or improved function into lasso peptides are highlighted, and unanswered questions in the field are also described.
Co-reporter:Si Jia Pan, Jakub Rajniak, Mikhail O. Maksimov and A. James Link
Chemical Communications 2012 vol. 48(Issue 13) pp:1880-1882
Publication Date(Web):16 Dec 2011
DOI:10.1039/C2CC17211A
The conserved threonine (Thr) residue in the penultimate position of the leader peptide of lasso peptides microcin J25 and capistruin can be effectively replaced by several amino acids close in size and shape to Thr. These findings suggest a model for lasso peptide biosynthesis in which the Thr sidechain is a recognition element for the lasso peptide maturation machinery.
Co-reporter:Siyan Zhang, Angel Long, and A. James Link
ACS Synthetic Biology 2012 Volume 1(Issue 3) pp:89
Publication Date(Web):September 26, 2011
DOI:10.1021/sb200002m
The Bcl-2 family of proteins regulates apoptosis at the level of mitochondrial permeabilization. Pro-death members of the family, including Bak and Bax, initiate apoptosis, whereas pro-survival members such as Bcl-xL and Mcl-1 antagonize the function of Bak and Bax via heterodimeric interactions. These heterodimeric interactions are primarily mediated by the binding of the helical amphipathic BH3 domain from a pro-death protein to a hydrophobic cleft on the surface of the pro-survival protein. Since high levels of pro-survival Bcl-2 proteins are present in many cancers, peptides corresponding to pro-death BH3 domains hold promise as therapeutics. Here we apply a high-throughput flow cytometry assay to engineer the Bak BH3 domain for improved affinity toward the pro-survival proteins Bcl-xL and Mcl-1. Two strategies, engineering the hydrophobic face of the Bak BH3 peptide and increasing its overall helicity, are successful in identifying Bak BH3 variants with improved affinity to Bcl-xL and Mcl-1. Hydrophobic face engineering of the Bak BH3 peptide led to variants with up to a 15-fold increase in affinity for Bcl-xL and increased specificity toward Bcl-xL. Engineering of the helicity of Bak BH3 led to modest (3- to 4-fold) improvements in affinity with retention of promiscuous binding to all pro-survival proteins. HeLa cell killing studies demonstrate that the affinity matured Bak BH3 variants retain their expected biological function.
Co-reporter:Siyan Zhang, Kiat Hwa Chan, Robert K. Prud'homme, and A. James Link
Molecular Pharmaceutics 2012 Volume 9(Issue 8) pp:2228-2236
Publication Date(Web):June 26, 2012
DOI:10.1021/mp3000748
Polymeric nanoparticles with multifunctional capabilities, including surface functionalization, hold great promise to address challenges in targeted drug delivery. Here, we describe a concise, robust synthesis of a heterofunctional polyethylene glycol (PEG), HO-PEG-azide. This macromer was used to synthesize polylactide (PLA)-PEG-azide, a functional diblock copolymer. Rapid precipitation of this copolymer with a hydrophobic cargo resulted in the generation of monodisperse nanoparticles with azides in the surface corona. To demonstrate conjugation to these nanoparticles, a regioselectively modified alkyne-folate was employed as a model small molecule ligand, and the artificial protein A1 with an alkyne moiety introduced by unnatural amino acid substitution was selected as a model macromolecular ligand. Using the copper-catalyzed azide–alkyne ligation reaction, both ligands exhibited good conjugation efficiency even when low concentrations of ligands were used.Keywords: bioconjugation; bioorthogonal reactions; click chemistry; nanoparticles;
Co-reporter:Si Jia Pan;Jakub Rajniak;Wai Ling Cheung; A. James Link
ChemBioChem 2012 Volume 13( Issue 3) pp:367-370
Publication Date(Web):
DOI:10.1002/cbic.201100596
Co-reporter:Diya M. Abdeljabbar;Hank J. Song
Biotechnology Letters 2012 Volume 34( Issue 1) pp:91-96
Publication Date(Web):2012 January
DOI:10.1007/s10529-011-0743-0
Cellulose degradation is essential for the future production of many advanced biofuels. Cellulases from the filamentous fungus Trichoderma reesei are among the most efficient enzymes for the hydrolysis of cellulosic materials. One of the cellulases from T. reesei, cellobiohydrolase II (CBH2), was studied because of its industrial relevance and proven enzymatic activity. Using both crude and rigorous membrane fractionation methods we show that full length T. reesei CBH2 is exclusively localized to the outer membrane when expressed recombinantly in Escherichia coli. Even fusing signal sequence-free maltose-binding protein to the N-terminus of CBH2, which has been shown to increase solubility of other proteins, did not prevent the outer membrane localization of CBH2. These results highlight the difficulties in producing fungal cellulases in bacterial hosts and provide a stepping stone for future cellulase engineering efforts.
Co-reporter:Mikhail O. Maksimov;István Pelczer
PNAS 2012 Volume 109 (Issue 38 ) pp:15223-15228
Publication Date(Web):2012-09-18
DOI:10.1073/pnas.1208978109
Lasso peptides are a class of ribosomally synthesized posttranslationally modified natural products found in bacteria. Currently
known lasso peptides have a diverse set of pharmacologically relevant activities, including inhibition of bacterial growth,
receptor antagonism, and enzyme inhibition. The biosynthesis of lasso peptides is specified by a cluster of three genes encoding
a precursor protein and two enzymes. Here we develop a unique genome-mining algorithm to identify lasso peptide gene clusters
in prokaryotes. Our approach involves pattern matching to a small number of conserved amino acids in precursor proteins, and
thus allows for a more global survey of lasso peptide gene clusters than does homology-based genome mining. Of more than 3,000
currently sequenced prokaryotic genomes, we found 76 organisms that are putative lasso peptide producers. These organisms
span nine bacterial phyla and an archaeal phylum. To provide validation of the genome-mining method, we focused on a single
lasso peptide predicted to be produced by the freshwater bacterium Asticcacaulis excentricus. Heterologous expression of an engineered, minimal gene cluster in Escherichia coli led to the production of a unique lasso peptide, astexin-1. At 23 aa, astexin-1 is the largest lasso peptide isolated to
date. It is also highly polar, in contrast to many lasso peptides that are primarily hydrophobic. Astexin-1 has modest antimicrobial
activity against its phylogenetic relative Caulobacter crescentus. The solution structure of astexin-1 was determined revealing a unique topology that is stabilized by hydrogen bonding between
segments of the peptide.
Co-reporter:Siyan Zhang, Robert K. Prud’homme, and A. James Link
Nano Letters 2011 Volume 11(Issue 4) pp:1723-1726
Publication Date(Web):March 21, 2011
DOI:10.1021/nl200271d
New sequencing technologies based on massively parallel signature sequencing (MPSS) have been developed to reduce the cost of genome sequencing. In some current MPSS platforms, DNA-modified micrometer-scale beads are used to template the polymerase chain reaction (PCR). Reducing the size of the beads to nanoscale can lead to significant improvements in sequencing throughput. To this end, we have assembled polymeric nanobeads that efficiently template PCR, resulting in DNA-decorated “nanobeads” with a high extent of functionalization.
Co-reporter:Si Jia Pan
Journal of the American Chemical Society 2011 Volume 133(Issue 13) pp:5016-5023
Publication Date(Web):March 10, 2011
DOI:10.1021/ja1109634
Microcin J25 (MccJ25) is a ribosomally synthesized antimicrobial peptide that has an unusual threaded lasso structure in which the C-terminal “tail” of the peptide is fed through a macrocyclic “ring” formed by the N-terminal residues. Production of MccJ25 in Escherichia coli is dependent upon a four-gene cluster encoding the structural gene mcjA, two maturation enzymes mcjB and mcjC, and an immunity factor, mcjD, in the form of an MccJ25 export pump. Here we have developed a system for orthogonal control of the expression of mcjA and mcjD, thus permitting independent control of MccJ25 production and export/immunity in E. coli. We used this system to screen saturation mutagenesis libraries targeted to either the ring or tail portions of MccJ25 and discovered nearly 100 new MccJ25 variants that retain antimicrobial function. While multiple amino acid substitutions in the tail portion of the peptide are well-tolerated, mutagenesis of the ring portion of the peptide is detrimental to the antimicrobial function of MccJ25. We demonstrated that the decreased function of the ring variants is due to the inability of these variants to be transported to the cytoplasm of susceptible strains. Additionally, we found several MccJ25 variants from the tail library with improved efficacy toward the MccJ25-sensitive strains E. coli and Salmonella enterica serovar Newport with the best variants exhibiting a nearly 5-fold increase in potency. The results described here provide further evidence that diverse amino acid sequences can be tolerated by the rigid lasso peptide fold.
Co-reporter:Siyan Zhang, Douglas H. Adamson, Robert K. Prud'homme and A. James Link
Polymer Chemistry 2011 vol. 2(Issue 3) pp:665-671
Publication Date(Web):20 Dec 2010
DOI:10.1039/C0PY00350F
The crosslinking of the core of nanoparticles composed of polystyrene-block-poly(ethylene oxide) copolymers can be achieved through encapsulation of a small molecule aryl diazide, 4,4′-diazidobiphenyl, and subsequent photolysis. The core-crosslinked nanoparticles exhibited high stability under thermal challenge. These stabilized nanoparticles have potential to serve as a nanobead for the polymerase chain reaction (PCR). We also demonstrated that this crosslinker can endow thin films of polystyrene with solvent resistance. NMR studies on these films provided evidence that crosslinking was occurring viainsertion of the nitrene formed by photolysis of the azide into the methylene or methine groups in the backbone of polystyrene.
Co-reporter:Siyan Zhang and A. James Link
Integrative Biology 2011 vol. 3(Issue 8) pp:823-831
Publication Date(Web):29 Jun 2011
DOI:10.1039/C1IB00023C
Members of the Bcl-2 family of proteins have opposing roles in programmed cell death; family members can play either pro-apoptotic or anti-apoptotic roles. Heterodimeric interactions between pro-apoptotic and anti-apoptotic members of the Bcl-2 family are critical for the regulation of apoptosis and are important targets for cancer therapeutics. Bcl-2 family interactions are mediated by the highly-conserved BH3 domain, corresponding to a single amphipathic α-helix, which binds in a hydrophobic cleft of its Bcl-2 family interaction partner. Here, using a high-throughput peptide-protein interaction assay based on bacterial cell surface display and flow cytometry, we present quantitative data for a near-complete set of 17 BH3 domains from the human genome binding to each of the 5 anti-apoptotic Bcl-2 family members. Biophysical insights into the affinity and specificity of these interactions are provided by analysis of the interactome data. In addition we carried out a truncation study of the Bim BH3 domain to define the core residues responsible for anti-apoptotic protein binding. The interactome data from this study has implications both in basic research on apoptosis and in the design of peptidic cancer therapeutics.
Co-reporter:Diya M. Abdeljabbar;Thomas J. Klein; A. James Link
ChemBioChem 2011 Volume 12( Issue 11) pp:1699-1702
Publication Date(Web):
DOI:10.1002/cbic.201100089
Co-reporter:Wai Ling Cheung ; Si Jia Pan
Journal of the American Chemical Society 2010 Volume 132(Issue 8) pp:2514-2515
Publication Date(Web):February 9, 2010
DOI:10.1021/ja910191u
The antimicrobial peptide microcin J25 (MccJ25) is matured by two enzymes, McjB and McjC, from a 58 amino acid (aa) preprotein, McjA, into its final 21 aa lasso topology. Herein we have investigated the role of the leader peptide of McjA and found that only the eight C-terminal amino acids of this leader peptide are required for maturation of MccJ25. There is a high content of lysine residues in the McjA leader peptide, but herein we also demonstrate that these charged amino acids do not play a major role in the maturation of MccJ25. Alanine scanning mutagenesis studies revealed that the Thr-35 residue in the leader peptide is critical for correct processing of McjA into mature MccJ25. In the absence of detailed structural and biochemical data about McjB and McjC, these studies allow us to propose a putative role for the leader peptide as a simple motif for docking of the McjA preprotein in the maturation enzymes.
Co-reporter:Diya M. Abdeljabbar ; Thomas J. Klein ; Siyan Zhang
Journal of the American Chemical Society 2009 Volume 131(Issue 47) pp:17078-17079
Publication Date(Web):November 6, 2009
DOI:10.1021/ja907969m
Engineered aminoacyl-tRNA synthetases have been used to enable the incorporation of many unnatural amino acids into recombinant proteins in vivo. In the majority of these studies, the engineered synthetase is harbored on a plasmid while the host retains a wild-type copy of the synthetase in its genome. Herein, we construct a strain carrying a single genomic copy of a methionyl-tRNA synthetase (MetRS) gene, metG*, engineered to enable the incorporation of azidonorleucine (ANL) into proteins. The resulting strain, M15MA metG*, is capable of both supporting robust cell growth and enabling the production of >20 mg/L culture of a recombinant protein, murine dihydrofolate reductase, containing ANL. The extent of replacement of methionine with ANL in this protein is 90%. Using this strain, we also produce ANL-containing OmpC, an outer membrane protein, and demonstrate that the surface of cells displaying this protein can be covalently modified using copper-catalyzed azide−alkyne cycloaddition. Since this mutant MetRS has been introduced into the genome, as opposed to a plasmid, M15MA metG* is genetically stable.
Co-reporter:Lucy Y. Xu
Biotechnology Letters 2009 Volume 31( Issue 11) pp:
Publication Date(Web):2009 November
DOI:10.1007/s10529-009-0075-5
The stress response of E. coli to the expression of two recombinant membrane proteins, the E. coli AAA+ protease FtsH and the human G-protein coupled receptor CB1, was examined using several members of a promoter-GFP library. Several genes from the heat-shock and envelope stress regulons (rpoH, clpP, lon, and ftsH) were strongly induced by expression of either membrane protein. Flow cytometry was used to monitor the real-time dynamics of the transcription of these reporter genes in response to membrane protein expression. Co-expression of CB1 and FtsH led to an additive response in these four reporter genes suggesting that the stresses may be occurring via different physiological mechanisms.
Co-reporter:
Nature Protocols 2007 2(8) pp:
Publication Date(Web):2007-07-26
DOI:10.1038/nprot.2007.268
Once shunned because of its perceived toxicity and reactivity, the azide functional group has emerged in the last 7 years as a significant facilitator of chemical biology and bioconjugation efforts1. As the azide moiety is essentially not present in nature, save for a single known natural product, it represents a nearly ideal bioorthogonal chemical handle. The use of the azide group in chemical biology was largely fueled by the discovery of chemical reactions that can specifically and rapidly form new bonds from azides under physiologically relevant conditions: the Staudinger ligation2, 3 and one of the click chemistry4 reactions, the copper-catalyzed azide–alkyne ligation (Fig. 1)5, 6. More recently, a variant of the azide–alkyne ligation that eliminates the need for a copper catalyst has been developed7. Azides have been introduced into several classes of biomolecules including sugars8, lipids9, proteins10 and nucleic acids11 and subsequently functionalized via the Staudinger ligation. The functionalization of larger biological structures is also possible as evidenced by the addition of fluorophores via azide–alkyne ligation on the surface of intact virus particles12.In 2002, in collaboration with the Bertozzi laboratory, we reported that AHA (2-amino-4-azidobutanoic acid; Fig. 2) serves as a surrogate for methionine in translation in auxotrophic Escherichia coli cells10. This amino acid, along with azidoalanine and azidonorvaline, had been previously prepared as a potential metabolite resulting from ionic azide treatment of bacteria13, 14. Although we have demonstrated that each of these amino acids along with the longer chain amino acid azidonorleucine can serve as surrogates for methionine in bacterial protein synthesis15, AHA is by far the most translationally active azide-bearing analog of methionine. The yield of purified AHA-substituted recombinant protein obtained in standard protein expression experiments is comparable to yields of wild-type, methionine-substituted protein. We have introduced AHA into the enzyme dihydrofolate reductase and the outer membrane protein OmpC with subsequent functionalization via the Staudinger ligation and azide–alkyne ligation, respectively10, 16. More recently, we have used a screen based on azide–alkyne ligation on the E. coli cell surface to facilitate the discovery of aminoacyl-tRNA synthetase (aaRS) mutants that permit high-level incorporation of azidonorleucine into recombinant proteins17.Because the incorporation of amino acids into proteins in vivo is governed largely by the host aaRS18, a convenient metric for the quality of surrogacy of a given non-canonical amino acid is its rate of activation by the corresponding aaRS. The catalytic efficiency (kcat/Km) of the E. coli methionyl-tRNA synthetase toward AHA is ca. 400-fold lower than that for methionine, and AHA is readily incorporated into proteins when cells are cultured in media depleted of methionine. In collaboration with Erin Schuman and colleagues, we have exploited the translational activity of AHA to develop the BONCAT (bioorthogonal non-canonical amino-acid tagging) method, a proteomic technology that permits direct interrogation of newly synthesized proteomes19. In BONCAT, AHA is introduced to cells concomitantly with a stimulus or some other challenge to the cell. Proteins synthesized subsequent to the stimulus or challenge are tagged with AHA, and these proteins are covalently labeled with a biotinylated affinity tag via azide–alkyne ligation. Following affinity purification on an avidin column, the newly synthesized proteome may be analyzed directly by any number of mass spectrometry techniques. A detailed protocol for BONCAT has previously been published in Nature Protocols20.In our laboratory, we have adopted two different approaches to synthesizing AHA. The first is a modified version of the original synthesis of Mangold et al. The second approach to synthesizing AHA is a more concise route developed in our laboratory, which relies on direct azidification of protected 2-aminobutanoic acid (Fig. 3). The first synthesis uses straightforward high-yielding synthetic transformations, but suffers from the necessity of having to protect the carboxylic acid moiety, adding extra protection and deprotection steps. The second synthesis is shorter, but the azidification reagent, triflic azide, must be prepared fresh before each synthesis. In this paper, we describe in detail the synthetic route to AHA developed in our laboratory, whereas a sister Nature Protocols paper by the same authors21 details our modified version of Mangold's original synthetic procedure.The synthetic route described herein is based on the direct azidification of amines in a copper-catalyzed reaction22. In this synthesis, Boc-protected diaminobutyric acid (Boc-Dab) is converted in one step to Boc-protected AHA by treatment with triflic azide (trifluoromethanesulfonic azide; see Fig. 3). The free amino acid is obtained upon treatment with acid and is purified by ion-exchange chromatography.Steps 1–11, preparation of triflic azide and azidification of Boc-Dab: 20 hSteps 12 and 13, deprotection of Boc-AHA: 1.5 hSteps 14–19, purification of cation-exchange resin: 3 hSteps 20–22, recrystallization of AHA: 16 hTo avoid excessive brown color of AHA after ion-exchange chromatography, perform additional washes of the Dowex resin with 1 N NH4OH.As described, the synthesis will produce 600–900 mg of AHA after ion-exchange chromatography or 400–700 mg following recrystallization. The procedure can be scaled up if required.AHA: 1H-NMR (D2O) δ 3.67 (m, 1H), 3.40 (t, 2H, J = 6.7 Hz), 1.97 (m, 2H).
Co-reporter:
Nature Protocols 2007 2(8) pp:
Publication Date(Web):2007-07-26
DOI:10.1038/nprot.2007.269
Please note that a more thorough discussion of the relevant background information and on the relative merits of the present approach to synthesizing AHA, in comparison to a competing method developed in our laboratory, can be found in a companion Nature Protocols paper by the same authors2, which specifically covers the mentioned alternative approach.Steps 1–6, protection of Boc-homoserine: 5 hSteps 7–14, tosylation of Boc-homoserine methyl ester: 10 hSteps 15–18, synthesis of Boc-AHA methyl ester: 16 hSteps 19–22, deprotection of AHA: 2 hSteps 23–28, purification of AHA by cation-exchange chromatography: 3 hSteps 29–31, recrystallization of AHA: 16 hTo avoid excessive brown color of AHA after ion-exchange chromatography, perform additional washes of the Dowex resin with 1 N NH4OH.As described, this synthesis will result in 500–700 mg of AHA. After recrystallization, this amount will be reduced to 300–500 mg.Tosylate of Boc-homoserine methyl ester: 1H-NMR (CDCl3) δ 7.79, 7.36 (m, 4H), 5.14 (m, 1H), 4.33 (m, 1H), 4.10 (t, 2H J = 6.3 Hz), 3.72 (s, 3H), 2.45 (s, 3H), 2.15 (m, 2H), 1.42 (s, 9H).Boc-AHA methyl ester: 1H-NMR (CDCl3) δ 5.20 (broad d, 1H), 4.40 (m, 1H), 3.76 (s, 3H), 3.41 (t, 2H, J = 6.9 Hz), 2.0 (m, 2H), 1.43 (s, 9H).AHA: 1H-NMR (D2O) δ 3.67 (m, 1H), 3.40 (t, 2H, J = 6.7 Hz), 1.97 (m, 2H).
Co-reporter:Si Jia Pan, Wai Ling Cheung, A. James Link
Protein Expression and Purification (June 2010) Volume 71(Issue 2) pp:200-206
Publication Date(Web):1 June 2010
DOI:10.1016/j.pep.2009.12.010
Microcin J25 (MccJ25) is an antimicrobial peptide produced by isolates of Escherichia coli with activity against closely related species. Production and export of mature MccJ25 in E. coli requires four genes that are organized on a plasmid-borne cluster in natural producer strains. In these strains, MccJ25 production does not commence until the cells reach stationary phase, and, according to previous literature, the highest titers of MccJ25 are obtained from cells grown in nutrient-poor medium. We sought to design an engineered MccJ25 gene cluster that alleviated the growth phase and media limitations of the natural cluster. In contrast to previous reports, we observe here that production of MccJ25 from its natural cluster is efficient in rich media, such as Luria–Bertani (LB). The engineered gene cluster functions in several E. coli strains and produces titers of MccJ25 that are moderately increased (1.5- to 2-fold) relative to the natural cluster. RT-PCR experiments and translational GFP fusion experiments confirm that the engineered cluster produces MccJ25 throughout exponential phase. Furthermore, we provide evidence that control of the natural MccJ25 gene cluster is at the transcriptional level. The observations herein provide design parameters for large-scale production of MccJ25 for biotechnological applications.
Co-reporter:Andrew L. Ferguson, Siyan Zhang, Igor Dikiy, Athanassios Z. Panagiotopoulos, Pablo G. Debenedetti, A. James Link
Biophysical Journal (3 November 2010) Volume 99(Issue 9) pp:
Publication Date(Web):3 November 2010
DOI:10.1016/j.bpj.2010.08.073
The antimicrobial peptide microcin J25 (MccJ25) is posttranslationally matured from a linear preprotein into its native lasso conformation by two enzymes. One of these enzymes cleaves the preprotein and the second enzyme installs the requisite isopeptide bond to establish the lasso structure. Analysis of a mimic of MccJ25 that can be cyclized without the influence of the maturation enzymes suggests that MccJ25 does not spontaneously adopt a near-lasso structure. In addition, we conducted atomistically detailed replica-exchange molecular dynamics simulations of pro-microcin J25 (pro-MccJ25), the 21-residue uncyclized analog of MccJ25, to determine the conformational ensemble explored in the absence of the leader sequence or maturation enzymes. We applied a nonlinear dimensionality reduction technique known as the diffusion map to the simulation trajectories to extract two global order parameters describing the fundamental dynamical motions of the system, and identify three distinct pathways. One path corresponds to the spontaneous adoption of a left-handed lasso, in which the N-terminus wraps around the C-terminus in the opposite sense to the right-handed topology of native MccJ25. Our computational and experimental results suggest a role for the MccJ25 leader sequence and/or its maturation enzymes in facilitating the adoption of the right-handed topology.
Co-reporter:Jingjing Sun, Diya M. Abdeljabbar, Nicole Clarke, Meghan L. Bellows, ... A. James Link
Journal of Molecular Biology (27 November 2009) Volume 394(Issue 2) pp:297-305
Publication Date(Web):27 November 2009
DOI:10.1016/j.jmb.2009.09.023
The interactions between pro- and anti-apoptotic members of the Bcl-2 class of proteins control whether a cell lives or dies, and the study of these protein–protein interactions has been an area of intense research. In this report, we describe a new tool for the study and engineering of apoptotic protein interactions that is based on the flow cytometric detection of these interactions on the surface of Escherichia coli. After validation of the assay with the well-studied interaction between the Bak(72–87) peptide and the anti-apoptotic protein Bcl-xL, the effect of both increasing and decreasing Bak peptide length on Bcl-xL binding was investigated. Previous work demonstrated that the Bak(72–87) peptide also binds to the anti-apoptotic protein Bcl-2, albeit with lower binding affinity compared to Bcl-xL. Here, we demonstrate that a slightly longer Bak peptide corresponding to amino acids 72–89 of Bak binds Bcl-xL and Bcl-2 equally well. Approximate binding affinity calculations on these peptide–protein complexes confirm the experimental observations. The flow cytometric assay was also used to screen a saturation mutagenesis library of Bak(72–87) variants for improved affinity to Bcl-xL. The best variants obtained from this library exhibit an apparent Kd to Bcl-xL 4-fold lower than that of wild-type Bak(72–87).
Co-reporter:Si Jia Pan, Jakub Rajniak, Mikhail O. Maksimov and A. James Link
Chemical Communications 2012 - vol. 48(Issue 13) pp:NaN1882-1882
Publication Date(Web):2011/12/16
DOI:10.1039/C2CC17211A
The conserved threonine (Thr) residue in the penultimate position of the leader peptide of lasso peptides microcin J25 and capistruin can be effectively replaced by several amino acids close in size and shape to Thr. These findings suggest a model for lasso peptide biosynthesis in which the Thr sidechain is a recognition element for the lasso peptide maturation machinery.
Co-reporter:Diya M. Abdeljabbar, Frank J. Piscotta, Siyan Zhang and A. James Link
Chemical Communications 2014 - vol. 50(Issue 94) pp:NaN14903-14903
Publication Date(Web):2014/10/09
DOI:10.1039/C4CC06528J
Here we demonstrate a methodology, termed protein stapling, for the introduction of covalent constraints into recombinant proteins. Using the azide–alkyne click reaction as the stapling chemistry, we have improved the thermostability of a model leucine zipper protein. Additionally, stapling the core of the small, globular protein G resulted in improved binding to its target, immunoglobulin G.
Co-reporter:Frank J. Piscotta, Jeffery M. Tharp, Wenshe R. Liu and A. James Link
Chemical Communications 2015 - vol. 51(Issue 2) pp:NaN412-412
Publication Date(Web):2014/11/04
DOI:10.1039/C4CC07778D
Using the amber suppression approach, four noncanonical amino acids (ncAAs) were used to replace existing amino acids at four positions in lasso peptide microcin J25 (MccJ25). The lasso peptide biosynthesis enzymes tolerated all four ncAAs and produced antibiotics with efficacy equivalent to wild-type in some cases. Given the rapid expansion of the genetically encoded ncAA pool, this study is the first to demonstrate an expedient method to significantly increase the chemical diversity of lasso peptides.

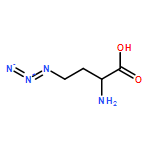
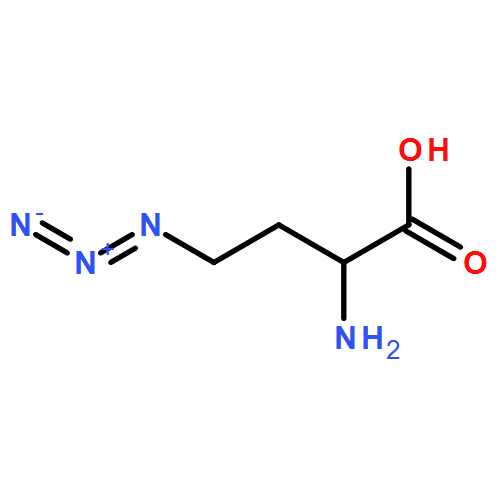
















![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3.png)
![4,5-Dioxo-4,5-dihydro-1H-pyrrolo[2,3-f]quinoline-2,7,9-tricarboxylic acid](/data/chemimg/654800/72909-34-3_b.png)