Co-reporter:Chih-Chung Tseng, Hannah Noordali, Monica Sani, Melanie Madhani, Denis M. Grant, Michael P. Frenneaux, Matteo Zanda, and Iain R. Greig
Journal of Medicinal Chemistry April 13, 2017 Volume 60(Issue 7) pp:2780-2780
Publication Date(Web):March 9, 2017
DOI:10.1021/acs.jmedchem.6b01592
We designed and synthesized perhexiline analogues that have the same therapeutic profile as the parent cardiovascular drug but lacking its metabolic liability associated with CYP2D6 metabolism. Cycloalkyl perhexiline analogues 6a–j were found to be unsuitable for further development, as they retained a pharmacokinetic profile very similar to that shown by the parent compound. Multistep synthesis of perhexiline analogues incorporating fluorine atoms onto the cyclohexyl ring(s) provided a range of different fluoroperhexiline analogues. Of these, analogues 50 (4,4-gem-difluoro) and 62 (4,4,4′,4′-tetrafluoro) were highly stable and showed greatly reduced susceptibility to CYP2D6-mediated metabolism. In vitro efficacy studies demonstrated that a number of derivatives retained acceptable potency against CPT-1. Having the best balance of properties, 50 was selected for further evaluation. Like perhexiline, it was shown to be selectively concentrated in the myocardium and, using the Langendorff model, to be effective in improving both cardiac contractility and relaxation when challenged with high fat buffer.
Co-reporter:Iain R. Greig, Gemma L. Baillie, Mostafa Abdelrahman, Laurent Trembleau, Ruth A. Ross
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 18) pp:4403-4407
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmcl.2016.08.018
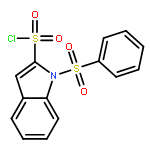
Existing CB1 negative allosteric modulators (NAMs) fall into a limited range of structural classes. In spite of the theoretical potential of CB1 NAMs, published in vivo studies have generally not been able to demonstrate the expected therapeutically-relevant CB1-mediated effects. Thus, a greater range of molecular tools are required to allow definitive elucidation of the effects of CB1 allosteric modulation. In this study, we show a novel series of indole sulfonamides. Compounds 5e and 6c (ABD1075) had potencies of 4 and 3 nM respectively, and showed good oral exposure and CNS penetration, making them highly versatile tools for investigating the therapeutic potential of allosteric modulation of the cannabinoid system.
Co-reporter:Iain R. Greig, Emmanuel Coste, Stuart H. Ralston, Robert J. van ’t Hof
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:816-820
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.11.067
Triaylsulfonamides were identified as novel anti-inflammatory agents, acting by inhibition of RANKL and TNFα signaling. Structure-activity studies led to the identification of compounds with in vitro potencies of <100 nM against J774 macrophages and osteoclasts, but with little activity against osteoblasts or hepatocytes (IC50 >50 μM). A representative compound (4k, ABD455) was able to completely prevent inflammation in vivo in a prevention model and was highly effective at controlling inflammation in a treatment model.
Co-reporter:Iain R. Greig, Emmanuel Coste, Stuart H. Ralston, Rob J. van’t Hof
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 18) pp:5548-5551
Publication Date(Web):15 September 2010
DOI:10.1016/j.bmcl.2010.07.055
Biphenylketones were identified as novel inhibitors of NFκB activation. Structure–activity studies led to the identification of compound 4c, which had good potency against osteoclasts (IC50 = 0.8 μM), showed oral activity, and was able to completely prevent inflammation and bone loss in vivo.

![3,4'-Difluoro-[1,1'-biphenyl]-4-carbonitrile](http://img.cochemist.com/ccimg/1214400/1214332-40-7.png)
![3,4'-Difluoro-[1,1'-biphenyl]-4-carbonitrile](http://img.cochemist.com/ccimg/1214400/1214332-40-7_b.png)







![TRIBUTYL-[CYCLOHEXYL(METHOXYMETHOXY)METHYL]STANNANE](/data/chemimg/364700/113248-99-0.png)
![TRIBUTYL-[CYCLOHEXYL(METHOXYMETHOXY)METHYL]STANNANE](/data/chemimg/364700/113248-99-0_b.png)
![2-Hexanone, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/110900/110862-03-8.png)
![2-Hexanone, 6-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/110900/110862-03-8_b.png)
![1,4-Dioxaspiro[4.5]decane-8-carbaldehyde](http://img.cochemist.com/ccimg/93300/93245-98-8.png)
![1,4-Dioxaspiro[4.5]decane-8-carbaldehyde](http://img.cochemist.com/ccimg/93300/93245-98-8_b.png)


![[1-(Phenylsulfonyl)-1H-indol-3-yl]methanol](http://img.cochemist.com/ccimg/89300/89241-33-8.png)
![[1-(Phenylsulfonyl)-1H-indol-3-yl]methanol](http://img.cochemist.com/ccimg/89300/89241-33-8_b.png)

![1,4-Dioxaspiro[4.5]dec-7-ene-8-carboxaldehyde](http://img.cochemist.com/ccimg/72500/72445-22-8.png)
![1,4-Dioxaspiro[4.5]dec-7-ene-8-carboxaldehyde](http://img.cochemist.com/ccimg/72500/72445-22-8_b.png)






![4'-Fluoro-[1,1'-biphenyl]-4-methanamine](http://img.cochemist.com/ccimg/776300/776291-03-3.png)
![4'-Fluoro-[1,1'-biphenyl]-4-methanamine](http://img.cochemist.com/ccimg/776300/776291-03-3_b.png)
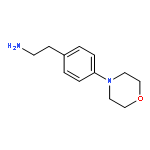
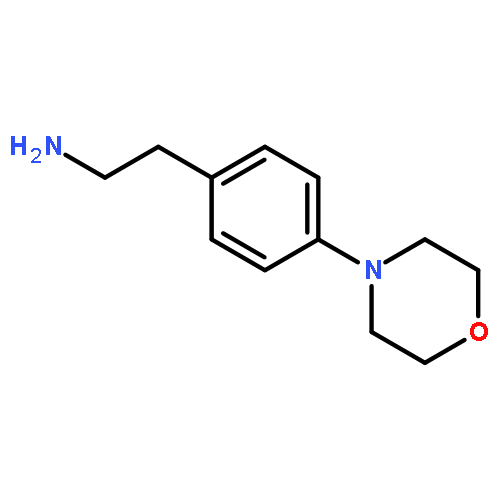
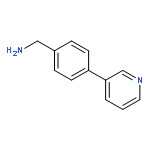
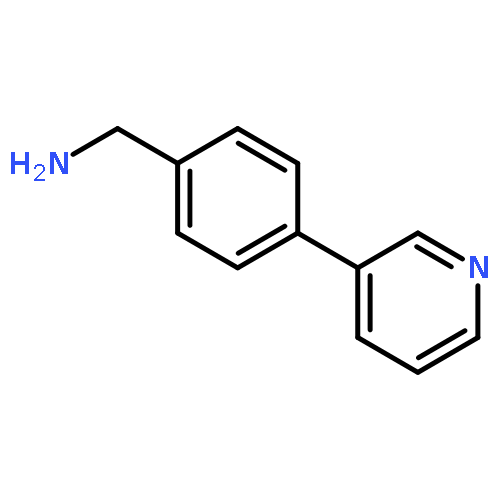












![Phenol, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-, rel-](/data/chemimg/1338600/83003-12-7.png)
![Phenol, 5-(1,1-dimethylheptyl)-2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-, rel-](/data/chemimg/1338600/83003-12-7_b.png)