Co-reporter:Chuan-Ming Wang;Yang-Dong Wang;Zai-Ku Xie
The Journal of Physical Chemistry C March 19, 2009 Volume 113(Issue 11) pp:4584-4591
Publication Date(Web):2017-2-22
DOI:10.1021/jp810350x
For its unique position in the coal chemical industry, the methanol to olefin (MTO) reaction has been a hot topic in zeolite catalysis. Due to the complexities of catalyst structure and reaction networks, many questions such as how the olefin chain is built from methanol remain elusive. On the basis of periodic density functional theory calculations, this work establishes the first complete catalytic cycle for MTO reaction via hexamethylbenzene (HMB) trapped in HSAPO-34 zeolite based on the so-called side chain hydrocarbon pool mechanism. The cycle starts from the methylation of HMB that leads to heptamethylbenzenium ion (heptaMB+) intermediate. This is then followed by the growth of side chain via repeated deprotonation of benzenium ions and methylation of the exocyclic double bond. Ethene and propene can finally be released from the side ethyl and isopropyl groups of benzenium ions by deprotonation and subsequent protonation steps. We demonstrate that (i) HMB/HSAPO-34 only yields propene as the primary product based on the side chain hydrocarbon pool mechanism and (ii) an indirect proton-shift step mediated by water that is always available in the system is energetically more favorable than the traditionally regarded internal hydrogen-shift step. Finally, the implications of our results toward understanding the effect of acidity of zeolite on MTO activity are also discussed.
Co-reporter:Yao-Ping Xie, Xiao-Jie Zhang, and Zhi-Pan Liu
Journal of the American Chemical Society February 22, 2017 Volume 139(Issue 7) pp:2545-2545
Publication Date(Web):February 6, 2017
DOI:10.1021/jacs.6b11193
Under mild static compression (15 GPa), graphite preferentially turns into hexagonal diamond, not cubic diamond, the selectivity of which is against thermodynamics. Here we, via novel potential energy surface global exploration, report seven types low energy intermediate structures at the atomic level that are key to the kinetics of graphite to diamond solid phase transition. On the basis of quantitative kinetics data, we show that hexagonal diamond has a facile initial nucleation mechanism inside graphite matrix and faster propagation kinetics owing to the presence of three coherent graphite/hexagonal diamond interfaces, forming coherent nuclei in graphite matrix. By contrast, for the lack of coherent nucleus core, the growth of cubic diamond is at least 40 times slower and its growth is inevitably mixing with that of hexagonal diamond.
Co-reporter:Dong Wang, Zhi-Pan Liu, and Wei-Min Yang
ACS Catalysis April 7, 2017 Volume 7(Issue 4) pp:2744-2744
Publication Date(Web):March 7, 2017
DOI:10.1021/acscatal.7b00225
Metal cocatalysts are widely utilized for enhancing photocatalytic conversion. In TiO2-based photocatalysts, a wide range of metals dispersed on TiO2 surfaces were observed to be effective for photocatalytic hydrogen production. To clarify the metal/oxide synergistic effect in photocatalysis and the insensitivity of photoactivity on metal types, here we investigate the mechanism of electron transfer from semiconductor to the cocatalyst by using ab initio molecular dynamics and hybrid density functional theory calculations. By determining the optimal geometry of a Pt13 subnano cluster on anatase TiO2(101) and quantifying the electron transfer energetics, we find that the electron transfer from oxide to the metal cluster is significantly boosted (exothermic by more than 0.3 eV) by the adsorption of proton on the metal cluster, which is otherwise endothermic without the presence of proton. This cooperative effect between oxide, subnano metal cluster, and adsorbed proton is rationalized from electronic structure analyses. We show that the proton-promoted electron transfer phenomenon in photocatalysis appears to be universally present, as evidenced from theoretical calculations by replacing Pt with other metals, including Co, Ni, Cu, Pd, and Rh. This mechanism differs fundamentally from the proton-coupled electron transfer frequently quoted in electrocatalysis and may assist the photocatalyst design toward highly efficient solar fuel production.Keywords: density functional theory; metal cocatalyst; photocatalytic hydrogen evolution; proton-coupled electron transfer; proton-promoted electron transfer; TiO2;
Co-reporter:Si-Da Huang;Cheng Shang;Xiao-Jie Zhang
Chemical Science (2010-Present) 2017 vol. 8(Issue 9) pp:6327-6337
Publication Date(Web):2017/08/21
DOI:10.1039/C7SC01459G
While the underlying potential energy surface (PES) determines the structure and other properties of a material, it has been frustrating to predict new materials from theory even with the advent of supercomputing facilities. The accuracy of the PES and the efficiency of PES sampling are two major bottlenecks, not least because of the great complexity of the material PES. This work introduces a “Global-to-Global” approach for material discovery by combining for the first time a global optimization method with neural network (NN) techniques. The novel global optimization method, named the stochastic surface walking (SSW) method, is carried out massively in parallel for generating a global training data set, the fitting of which by the atom-centered NN produces a multi-dimensional global PES; the subsequent SSW exploration of large systems with the analytical NN PES can provide key information on the thermodynamics and kinetics stability of unknown phases identified from global PESs. We describe in detail the current implementation of the SSW-NN method with particular focuses on the size of the global data set and the simultaneous energy/force/stress NN training procedure. An important functional material, TiO2, is utilized as an example to demonstrate the automated global data set generation, the improved NN training procedure and the application in material discovery. Two new TiO2 porous crystal structures are identified, which have similar thermodynamics stability to the common TiO2 rutile phase and the kinetics stability for one of them is further proved from SSW pathway sampling. As a general tool for material simulation, the SSW-NN method provides an efficient and predictive platform for large-scale computational material screening.
Co-reporter:Xiao-Jie Zhang;Cheng Shang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 6) pp:4725-4733
Publication Date(Web):2017/02/08
DOI:10.1039/C6CP06895B
The crystal to amorphous transformation is a common phenomenon in Nature and has important impacts on material properties. Our current knowledge on such complex solid transformation processes is, however, limited because of their slow kinetics and the lack of long-range ordering in amorphous structures. To reveal the kinetics in the amorphization of solids, this work, by developing iterative reaction sampling based on the stochastic surface walking global optimization method, investigates the well-known crystal to amorphous transformation of silica (SiO2) under external pressures, the mechanism of which has long been debated for its non-equilibrium, pressure-sensitive kinetics and complex product components. Here we report for the first time the global potential energy surface (PES) and the lowest energy pathways for α-quartz amorphization from first principles. We show that the pressurization at 15 GPa, the reaction condition, can lift the quartz phase energetically close to the amorphous zone, which thermodynamically initializes the amorphization. More importantly, the large flexibility of Si cation coordination (including four, five and six coordination) results in many kinetically competing routes to more stable dense forms, including the known MI, stishovite, newly-identified MII and TI phases. All these pathways have high barriers due to the local Si–O bond breaking and are mediated by amorphous structures with five-fold Si. This causes simultaneous crystal-to-crystal and crystal-to-amorphous transitions. The high barrier and the reconstructive nature of the phase transition are the key kinetics origin for silica amorphization under pressures.
Co-reporter:Cheng Shang;Xiao-Jie Zhang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 47) pp:32125-32131
Publication Date(Web):2017/12/06
DOI:10.1039/C7CP07060H
Because of their weak intermolecular forces and flexible molecular geometry, molecular crystals are renowned for their structural versatility (polymorphism) and the great difficulty in controlling the crystal form during synthesis. Despite its great importance in determining the final solid form (e.g. single crystal, polycrystal or amorphous), the kinetics of the crystal-to-crystal transformation between structures with different molecular packing has long been a fundamental challenge in both measurement and simulation. Here we report the first global potential energy surface (PES) for urea crystals obtained by stochastic surface walking global PES exploration. With the big data from thousands of crystal/amorphous forms, we, using exhaustive reaction pathway sampling, resolve the solid-to-solid transformation pathways between urea crystals from first principles. We demonstrate that the strong tendency to grow a large single crystal of urea can be attributed to the flat PES between major crystal forms that share the same hydrogen-bonding network pattern, where one crystal can transform to another facilely via crystal-to-crystal transition. Other crystal forms with distinct hydrogen-bonding network patterns can be excluded in crystallization due to their poor thermodynamic stability and high barrier of solid-to-solid transition. A general theory for predicting molecular solid transformation is proposed and illustrated in a simplified one-dimensional global PES, which is now obtainable from computational techniques established here.
Co-reporter:Limin Zhang, Fangling Liu, Xuemei Sun, Guang-feng Wei, Yang TianZhi-pan Liu, Rong Huang, Yanyan Yu, Huisheng Peng
Analytical Chemistry 2017 Volume 89(Issue 3) pp:
Publication Date(Web):January 5, 2017
DOI:10.1021/acs.analchem.6b04168
Ascorbic acid (AA) levels are closely correlated with physiological and pathological events in brain diseases, but the mechanism remains unclear, mainly due to the difficulty of accurately analyzing AA levels in live brain. In this study, by engineering tunable defects and oxygen-containing species in carbon nanotubes, a novel aligned carbon nanotube fiber was developed as an accurate microsensor for the ratiometric detection of AA levels in live rat brains with Alzheimer’s disease (AD). AA oxidation is greatly facilitated on the fiber surface at a low potential, leading to high sensitivity as well as high selectivity against potential sources of interference in the brain. Additionally, an unexpected, separate peak from the fiber surface remains constant as the AA concentration increases, enabling real-time and ratiometric detection with high accuracy. The results demonstrated that the AA levels were estimated to be 259 ± 6 μM in cortex, 264 ± 20 μM in striatum, and 261 ± 21 μM in hippocampus, respectively, in normal condition. However, the overall AA level was decreased to 210 ± 30 μM in cortex, 182 ± 5 μM in striatum, and 136 ± 20 μM in hippocampus in the rat brain model of AD. To the best of our knowledge, this work is the first to accurately detect AA concentrations in the brains of live animal model of AD.
Co-reporter:Ye-Fei Li; Sheng-Cai Zhu
Journal of the American Chemical Society 2016 Volume 138(Issue 16) pp:5371-5379
Publication Date(Web):April 7, 2016
DOI:10.1021/jacs.6b01768
As a model system of 2-D oxide material, layered δ-MnO2 has important applications in Li ion battery systems. δ-MnO2 is also widely utilized as a precursor to synthesize other stable structure variants in the MnO2 family, such as α-, β-, R-, and γ-phases, which are 3-D interlinked structures with different tunnels. By utilizing the stochastic surface walking (SSW) pathway sampling method, we here for the first time resolve the atomistic mechanism and the kinetics of the layer-to-tunnel transition of MnO2, that is, from δ-MnO2 to the α-, β-, and R-phases. The SSW sampling determines the lowest-energy pathway from thousands of likely pathways that connects different phases. The reaction barriers of layer-to-tunnel phase transitions are found to be low, being 0.2–0.3 eV per formula unit, which suggests a complex competing reaction network toward different tunnel phases. All the transitions initiate via a common shearing and buckling movement of the MnO2 layer that leads to the breaking of the Mn–O framework and the formation of Mn3+ at the transition state. Important hints are thus gleaned from these lowest-energy pathways: (i) the large pore size product is unfavorable for the entropic reason; (ii) cations are effective dopants to control the kinetics and selectivity in layer-to-tunnel transitions, which in general lowers the phase transition barrier and facilitates the creation of larger tunnel size; (iii) the phase transition not only changes the electronic structure but also induces the macroscopic morphology changes due to the interfacial strain.
Co-reporter:Guang-Feng Wei and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 9) pp:4698-4706
Publication Date(Web):August 2, 2016
DOI:10.1021/acs.jctc.6b00556
Subnano transition metal particles have wide applications in chemistry. For the complexity of their potential energy surface, it has long been a great challenge for both theory and experiment to determine the structure of subnano clusters and thus predict their physiochemical properties. Here we explore the structure configurations for 35 subnano PtN (N = 12–46) clusters using a first-principles Stochastic Surface Walking (SSW) global search. For each cluster, thousands of structure candidates are collected from a parallel SSW search. This leads to the finding of 20 new global minima in 35 clusters, which reflects the essence of a first-principles global search for revealing the structure of subnano transition metal clusters. PtN subnano clusters with N being 14, 18, 22, 27, 36, and 44 have higher stability than their neighboring size clusters and are characterized as magic number clusters. These PtN subnano clusters exhibit metallic characteristics with a diminishing HOMO–LUMO gap, much poorer binding energy (by 1–1.7 eV), and a much higher Fermi level (by 1–1.5 eV) than bulk metal, implying their high chemical activity. By analyzing their structures, we observe the presence of a rigid core and a soft shell for PtN clusters and find that the core–shell 3-D architecture evolves as early as N > 22. For these core–shell clusters, a good core–shell lattice match is the key to achieve the high stability.
Co-reporter:Sheng-Cai Zhu, Shu-Hui Guan and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 27) pp:18563-18574
Publication Date(Web):20 Jun 2016
DOI:10.1039/C6CP03673B
Solid-to-solid phase transition, although widely exploited in making new materials, challenges persistently our current theory for predicting its complex kinetics and rich microstructures in transition. The Ga2O3 α–β phase transformation represents such a common but complex reaction with marked change in cation coordination and crystal density, which was known to yield either amorphous or crystalline products under different synthetic conditions. Here we, via recently developed stochastic surface walking (SSW) method, resolve for the first time the atomistic mechanism of Ga2O3 α–β phase transformation, the pathway of which turns out to be the first reaction pathway ever determined for a new type of diffusionless solid phase transition, namely, pseudomartensitic phase transition. We demonstrate that the sensitivity of product crystallinity is caused by its multi-step, multi-type reaction pathway, which bypasses seven intermediate phases and involves all types of elementary solid phase transition steps, i.e. the shearing of O layers (martensitic type), the local diffusion of Ga atoms (reconstructive type) and the significant lattice dilation (dilation type). While the migration of Ga atoms across the close-packed O layers is the rate-determining step and yields “amorphous-like” high energy intermediates, the shearing of O layers contributes to the formation of coherent biphase junctions and the presence of a crystallographic orientation relation, (001)α//(20)β + [120]α//[12]β. Our experiment using high-resolution transmission electron microscopy further confirms the theoretical predictions on the atomic structure of biphase junction and the formation of (20)β twin, and also discovers the late occurrence of lattice expansion in the nascent β phase that grows out from the parent α phase. By distinguishing pseudomartensitic transition from other types of mechanisms, we propose general rules to predict the product crystallinity of solid phase transition. The new knowledge on the kinetics of pseudomartensitic transition complements the theory of diffusionless solid phase transition.
Co-reporter:Shu-Hui Guan and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 6) pp:4527-4534
Publication Date(Web):12 Jan 2016
DOI:10.1039/C5CP07299A
Structural inhomogeneity is ubiquitous in solid crystals and plays critical roles in phase nucleation and propagation. Here, we develop a heterogeneous solid–solid phase transition theory for predicting the prevailing heterophase junctions, the metastable states governing microstructure evolution in solids. Using this theory and first-principles pathway sampling simulation, we determine two types of heterophase junctions pertaining to metal α–ω phase transition at different pressures and predict the reversibility of transformation only at low pressures, i.e. below 7 GPa. The low-pressure transformation is dominated by displacive Martensitic mechanism, while the high-pressure one is controlled by the reconstructive mechanism. The mechanism of α–ω phase transition is thus highly pressure-sensitive, for which the traditional homogeneous model fails to explain the experimental observations. The results provide the first atomic-level evidence on the coexistence of two different solid phase transition mechanisms in one system.
Co-reporter:Shu-Hui Guan, Xiao-Jie Zhang, and Zhi-Pan Liu
The Journal of Physical Chemistry C 2016 Volume 120(Issue 43) pp:25110-25116
Publication Date(Web):October 17, 2016
DOI:10.1021/acs.jpcc.6b08942
Solid-to-solid phase transition, although widely encountered in Nature, challenges persistently our current theory for predicting its complex kinetics and rich microstructures in transition. As an important representative for A2BO4 oxide, olivine (Mg2SiO4) to spinel solid phase transition was hotly debated for its unusual kinetics with complex cation and anion movement. Here we, via novel first-principles based global potential energy surface sampling, resolve the potential energy surface of Mg2SiO4 and the lowest energy pathway of the olivine-spinel solid phase transition. Instead of the traditionally regarded mechanism via shearing close-packing oxygen lattice, we identify an unprecedented pathway involving silicate oxoanion (SiO44–) rigid-body rotation and translation in the reaction, which may only be expected in molecular crystal transformation. While the anions and cations move synchronously, it is the coordination change of Mg2+ in reaction, from octahedral to tetrahedral sites that cause the high barrier. The replacement of Mg cation by Fe and Zn cations is found to substantially reduce the barrier of crystal transformation. Although the reaction do involve significant shearing with a [001](010) slip system, leading to the seemingly early formation of O sublattice, our theoretical results suggest that the phase transition is governed by incoherent nucleation and growth due to the lack of coherent heterophase interfaces. The theoretical results are of general importance for understanding the stability and phase transition mechanism in silicate group materials.
Co-reporter:Shu-Hui Guan; Xiao-Jie Zhang
Journal of the American Chemical Society 2015 Volume 137(Issue 25) pp:8010-8013
Publication Date(Web):June 15, 2015
DOI:10.1021/jacs.5b04528
The solid-phase transitions of zirconia are important phenomena for many industrial applications. Because of the lack of tools for resolving the atom displacement pattern, the transition kinetics has been disputed for over 60 years. Here, first-principles-based stochastic surface walking (SSW) pathway sampling is utilized for resolving the mechanism of ZrO2 tetragonal-to-monoclinic solid-phase transition. Two types of lattice and atom correspondence allowed in phase transition are determined for the first time from energy criterion, which are originated from two nearly energy-degenerate lowest-transition pathways and one stress-induced ferroelastic transition channel of tetragonal phase. An orthorhombic crystal phase (Pbc2/1) is discovered to be a trapping state at low temperatures in phase transition, the presence of which does not create new orientation relation but deters transformation toughening significantly. This new finding may facilitate the design of new functional oxide materials in ceramic industry.
Co-reporter:Sheng-Cai Zhu; Song-Hai Xie
Journal of the American Chemical Society 2015 Volume 137(Issue 35) pp:11532-11539
Publication Date(Web):August 19, 2015
DOI:10.1021/jacs.5b07734
The solid phase transition of TiO2, in particular anatase to rutile, has been extensively studied in the past 30 years. To seek the nucleation site at the beginning of phase transition is highly challenging, which asks for new theoretical techniques with high spatial and temporal resolution. This work reports the first evidence on the atomic structure of the nucleation sites in the TiO2 anatase-to-rutile phase transition. Novel automated theoretical methods, namely stochastic surface walking based pathway sampling methods, are utilized to resolve the lowest energy pathways at the initial stage of phase transition. We show that among common anatase surfaces, only the (112) ridged surface provides the nucleation site for phase transition, which can lead to the formation of both TiO2-II and brookite thin slabs. The TiO2-II phase is kinetically preferred product; the propagation into the subsurface is still hindered by high barriers that is the origin for the slow kinetics of nuclei formation. The rutile nuclei are thus not rutile phase but nascent metastable TiO2-II phase in an anatase matrix. The phase transition kinetics is found to be sensitive to the compressive strain and the crystallographic directions. The results rationalize the size and morphology dependence of the anisotropic phase transition kinetics of anatase particles and could facilitate the rational design of material via controlled solid phase transition.
Co-reporter:Wei-Na Zhao, Sheng-Cai Zhu, Ye-Fei Li and Zhi-Pan Liu
Chemical Science 2015 vol. 6(Issue 6) pp:3483-3494
Publication Date(Web):07 Apr 2015
DOI:10.1039/C5SC00621J
The heterophase solid–solid junction as an important type of structure unit has wide applications for its special mechanics and electronic properties. Here we present a first three-phase atomic model for the anatase–rutile TiO2 heterophase junction and determine its optical and electronic properties, which leads to resolution of the long-standing puzzles on the enhanced photocatalytic activity of anatase–rutile photocatalysts. By using a set of novel theoretical methods, including crystal phase transition pathway sampling, interfacial strain analysis and first principles thermodynamics evaluation of holes and electrons, we identify an unusual structurally ordered three-phase junction, a layer-by-layer “T-shaped” anatase/TiO2-II/rutile junction, for linking anatase with rutile. The intermediate TiO2-II phase, although predicted to be only a few atomic layers thick in contact with anatase, is critical to alleviate the interfacial strain and to modulate photoactivity. We demonstrate that the three-phase junction acts as a single-way valve allowing the photogenerated hole transfer from anatase to rutile but frustrating the photoelectron flow in the opposite direction, which otherwise cannot be achieved by an anatase–rutile direct junction. This new model clarifies the roles of anatase, rutile and the phase junction in achieving high photoactivity synergistically and provides the theoretical basis for the design of better photocatalysts by exploiting multi-phase junctions.
Co-reporter:Guang-Feng Wei and Zhi-Pan Liu
Chemical Science 2015 vol. 6(Issue 2) pp:1485-1490
Publication Date(Web):26 Nov 2014
DOI:10.1039/C4SC02806F
The restructuring of nanoparticles at the in situ condition is a common but complex phenomenon in nanoscience. Here, we present the first systematic survey on the structure dynamics and its catalytic consequence for hydrogen evolution reaction (HER) on Pt nanoparticles, as represented by a magic number Pt44 octahedron (∼1 nm size). Using a first principles calculation based global structure search method, we stepwise follow the significant nanoparticle restructuring under HER conditions as driven by thermodynamics to expose {100} facets, and reveal the consequent large activity enhancement due to the marked increase of the concentration of the active site, being identified to be apex atoms. The enhanced kinetics is thus a “byproduct” of the thermodynamical restructuring. Based on the results, the best Pt catalyst for HER is predicted to be ultrasmall Pt particles without core atoms, a size below ∼20 atoms.
Co-reporter:Xiao-Jie Zhang and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4885-4894
Publication Date(Web):September 25, 2015
DOI:10.1021/acs.jctc.5b00641
To identify the low energy pathway for solid-to-solid phase transition has been a great challenge in physics and material science. This work develops a new theoretical method, namely, variable-cell double-ended surface walking (VC-DESW) to locate the transition state (TS) and deduce the pathway in solid phase transition. Inherited from the DESW method ( J. Chem. Theory Comput. 2013, 9, 5745) for molecular systems, the VC-DESW method implements an efficient mechanism to couple the lattice and atom degrees of freedom. The method features with fast pseudopathway building and accurate TS location for solid phase transition systems without requiring expensive Hessian computation and iterative pathway optimization. A generalized coordinate, consisting of the lattice vectors and the scaled atomic coordinates, is designed for describing the crystal potential energy surface (PES), which is able to capture the anisotropic behavior in phase transition. By comparing with the existing method for solid phase transition in different systems, we show that the VC-DESW method can be much more efficient for finding the TS in crystal phase transition. With the combination of the recently developed unbiased stochastic surface walking pathway sampling method, the VC-DESW is further utilized to resolve the lowest energy pathway of SiO2 α-quartz to quartz-II phase transition from many likely reaction pathways. These new methods provide a powerful platform for understanding and predicting the solid phase transition mechanism and kinetics.
Co-reporter:Xiao-Jie Zhang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 4) pp:2757-2769
Publication Date(Web):01 Dec 2014
DOI:10.1039/C4CP04456H
The prediction of chemical reactivity and thus the design of new reaction systems are the key challenges in chemistry. Here, we develop an unbiased general-purpose reaction sampling method, the stochastic surface walking based reaction sampling (SSW-RS) method, and show that the new method is a promising solution for reactivity prediction of complex reaction systems. The SSW-RS method is capable of sampling both the configuration space of the reactant and the reaction space of pathways, owing to the combination of two recently developed theoretical methods, namely, the stochastic surface walking (SSW) method for potential energy surface (PES) exploration and the double-ended surface walking (DESW) method for building pathways. By integrating with first principles calculations, we show that the SSW-RS method can be applied to investigate the kinetics of complex organic reactions featuring many possible reaction channels and complex hydrogen-bonding networks, as demonstrated here using two examples, epoxypropane hydrolysis in aqueous solution and β-D-glucopyranose decomposition. Our results show that simultaneous sampling of the soft hydrogen-bonding conformations and the chemical reactions involving hard bond making/breaking can be achieved in the SSW-RS simulation, and the mechanism and kinetics can be predicted without a priori information on the system. Unexpected new chemistry for these reactions is revealed and discussed. In particular, despite many possible pathways for β-D-glucopyranose decomposition, the SSW-RS shows that only β-D-glucose and levoglucosan are kinetically preferred direct products and the 5- or 7-member ring products should be secondary products derived from β-D-glucose or levoglucosan. As a general tool for reactivity prediction, the SSW-RS opens a new route for the design of rational reactions.
Co-reporter:Guang-Feng Wei, Cheng Shang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 3) pp:2078-2087
Publication Date(Web):19 Nov 2014
DOI:10.1039/C4CP04145C
A supported nanoparticle is dynamic in reaction conditions, but how this dynamic behavior is influenced by the support remains elusive. Using the stochastic surface walking global optimization method, herein, we report the structure, thermodynamics and catalytic properties of Pt nanoparticles inside and outside a carbon nanotube (CNT). We reveal that confined metal nanoparticles are, surprisingly, structurally more flexible at low temperatures but less likely to melt at high temperatures. By investigating the O2 interaction with a Pt15 cluster inside and outside the CNT, we find that the low temperature structure versatility facilitates the in situ creation of favorable reaction sites, and thus maintains the catalytic activity of O2 dissociation. The decrease of the population for the liquid-like structures (largely disordered) offers the higher stability of the confined nanocatalyst. The theoretical results are consistent with experimental findings for the enhanced antioxidation ability of the confined metal nanoparticles.
Co-reporter:Ya-Hui Fang, Zhi-Pan Liu
Surface Science 2015 Volume 631() pp:42-47
Publication Date(Web):January 2015
DOI:10.1016/j.susc.2014.05.014
•A theoretical model to predict Tafel kinetics of MOR is established.•The free energy profiles of the C-H and O-H pathways are calculated.•The rate-determining step of methanol oxidation is the first C-H bond breaking.•The charge transfer coefficient and Tafel slope of MORare determined.Electrocatalytic methanol oxidation is of fundamental importance in electrochemistry and also a key reaction in direct methanol fuel cell. To resolve the kinetics at the atomic level, this work investigates the potential-dependent reaction kinetics of methanol oxidation on Pt(111) using the first principles periodic continuum solvation model based on modified-Poisson–Boltzmann equation (CM-MPB), focusing on the initial dehydrogenation elementary steps. A theoretical model to predict Tafel kinetics (current vs potential) is established by considering that the rate-determining step of methanol oxidation (to CO) is the first CH bond breaking (CH3OH(aq) → CH2OH* + H*) according to the computed free energy profile. The first CH bond breaking reaction needs to overcome a large entropy loss during methanol approaching to the surface and replacing the adsorbed water molecules. While no apparent charge transfer is involved in this elementary step, the charge transfer coefficient of the reaction is calculated to be 0.36, an unconventional value for charge transfer reactions, and the Tafel slope is deduced to be 166 mV. The results show that the metal/adsorbate interaction and the solvation environment play important roles on influencing the Tafel kinetics. The knowledge learned from the potential-dependent kinetics of methanol oxidation can be applied in general for understanding the electrocatalytic reactions of organic molecules at the solid–liquid interface.
Co-reporter:Sheng-Cai Zhu;Shu-Hui Guan;Wei-Na Zhao
Topics in Catalysis 2015 Volume 58( Issue 10-11) pp:644-654
Publication Date(Web):2015 August
DOI:10.1007/s11244-015-0410-0
The heterophase junction is of great importance in photocatalysis and many other applications. The mechanical, optical and electrical properties of metrical are sensitive to the atomic structure of the heterophase junction. To date, the determination of these structures at the atomic level has been of great challenge for both experiment and theory. In this focused review, we introduce the recently developed theoretical methods based on stochastic surface walking method, which in combination with finite strain theory and first principles calculations can be utilized for identifying the homogeneous phase transition pathway and characterizing the atomic structure at the phase junction. The orientation relation and the atomic habit plane obtained from theory provide the key information for constructing the coherent phase junction. We then discuss the application of the method in two example systems in the context of latest experimental findings, namely TiO2-B/anatase and anatase/rutile heterophase junction that are of wide application in photocatalysis. Using these examples, the merits and deficiencies of the current theoretical tools are illustrated. New theoretical approaches are called for towards the simulation of the solid-to-solid phase transition in real time and the characterization of heterostructure junction in general.
Co-reporter:Dr. Hongying Zhao;Dr. Guangfeng Wei;Dr. Junxia Gao;Dr. Zhipan Liu;Dr. Guohua Zhao
ChemElectroChem 2015 Volume 2( Issue 3) pp:366-373
Publication Date(Web):
DOI:10.1002/celc.201402372
Abstract
An ultrasound-enhanced electrochemical oxidation process with boron-doped diamond anodes is adopted and used for removing bisphenol A. The reaction activity and mechanism is evaluated and explained through experimental and theoretical methods. The structure and stability of the anode surface are illustrated and the electrochemical oxidation channels leading to ⋅OH formation are proposed using density functional calculations. Ultrasound increases the diffusion and mass transfer of ⋅OH at a high working potential (i.e., 3.0 V), while enhancing the collision frequency between organic pollutants and *O radicals at low working potentials (i.e., 1.7 V). The main intermediates determined by HPLC are hydroquinone, maleic and oxalic acids in both processes, suggesting the same degradation pathway even with ultrasound.
Co-reporter:Ya-Hui Fang and Zhi-Pan Liu
ACS Catalysis 2014 Volume 4(Issue 12) pp:4364
Publication Date(Web):October 28, 2014
DOI:10.1021/cs501312v
The Tafel equation is of fundamental importance in electrochemical kinetics, formulating a quantitative relation between the current and the applied electrochemical potential. The recent years have seen the rapid expansion and development in the application of first-principles density functional theory (DFT) simulation on electrocatalytic reactions that occur at the solid–liquid interface. This article reviews the current theoretical methods for electrochemistry modeling, in particular, those for the direct computation of Tafel kinetics of electrocatalytic reactions on surfaces based on DFT calculations. Representative reactions, namely, hydrogen evolution and oxygen evolution reactions, are selected to illustrate how the theoretical methods are applied to compute quantitatively the kinetics of multiple-step electrochemical reactions. We summarize in detail the computation procedure based on the first-principles periodic continuum solvation method for obtaining the charge transfer coefficient (CTC) and deducing the potential-dependent reaction rate. The theoretical results on the Tafel kinetics of electrochemical reactions are generalized and discussed.Keywords: charge transfer coefficient; electrocatalytic reactions; first-principles calculations; periodic continuum solvation method; Tafel kinetics
Co-reporter:Wei-Na Zhao and Zhi-Pan Liu
Chemical Science 2014 vol. 5(Issue 6) pp:2256-2264
Publication Date(Web):13 Feb 2014
DOI:10.1039/C3SC53385A
Photocatalytic water splitting is regarded as an important route for generating renewable energy. Here, charged-slab first principles calculations integrated with a periodic continuum solvation model is utilized to analyze the initiating steps of water splitting on the two most common TiO2 surfaces, namely, rutile (110) and anatase (101), at the solid–water interface. It is found that the first proton removal of water (H2O + hole+ → OH + H+) is sensitive to the crystalline phase and surface. The rutile (110) surface is more active for water splitting, with the calculated barrier of O–H bond breaking being 0.2 eV lower compared to that on anatase (101). The higher activity of rutile is not due to the redox level of the hole (the position of the valence band maximum), but caused by the more favorable local bonding geometry of the surface. Unexpectedly, the photogenerated hole does not promote O–H bond breaking, and the charge transfer occurs after the H2O dissociation when the surface O nearby the dissociated OH anion traps the hole. The solvation plays an important catalytic role to stabilize and remove protons from the reaction site, which effectively inhibits the charge-recombination of the dissociated OH anion with the proton. The theory presented here shows that the chemical properties of the surface play a significant role in the photocatalytic process, and a strategy based on simple structural parameters is proposed towards the design of new photocatalysts.
Co-reporter:Cheng Shang, Xiao-Jie Zhang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 33) pp:17845-17856
Publication Date(Web):12 Jun 2014
DOI:10.1039/C4CP01485E
The determination of crystal structures and the solid-to-solid phase transition mechanisms are two important and related subjects in material science. Here we develop an unbiased general-purpose potential energy surface (PES) searching method, namely, SSW-crystal method, for prediction of both the crystal structure and the crystal phase transition pathway. The SSW-crystal method features with stochastic surface walking (SSW) via repeated small structural perturbation by taking into account the second derivative information on both the lattice and the atom degrees of freedom. The SSW-crystal method is capable of overcoming the high barrier of phase transition and identifying the desirable phase transition reaction coordinates. By applying the SSW-crystal method to a set of examples, including SiO2 crystal up to 162 atoms per cell, Lennard-Jones model crystals up to 256 atoms, ternary SrTiO3 crystal of 50 atoms and the rutile-to-anatase TiO2 phase transition, we show that the SSW-crystal method can efficiently locate the global minimum (GM) from random initial structures without a priori knowledge of the system, and also allows for exhaustive sampling of the phase transition pathways, from which the lowest energy pathway can be obtained.
Co-reporter:Jing Sun, Ya-Hui Fang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 27) pp:13733-13740
Publication Date(Web):27 Feb 2014
DOI:10.1039/C4CP00037D
The Fe/N/C catalysts have emerged recently as a representative class of non-Pt catalysts for oxygen electrocatalytic reduction, which could have a competitive catalytic performance to Pt. However, the nature of the catalyst remains elusive, especially on the active site structure and the electrocatalytic kinetics. Here we examine two kinds of Fe/N active sites for Fe/N/C catalysts, namely, the four-coordinated FeN4 and the five-coordinated Fe(CN)N4 centers embedded in graphene layers. By using large-scale first principles calculations with a periodic continuum solvation model based on the Modified-Poisson–Boltzmann equation (CM-MPB), we identified the four (4e) and two electron (2e) oxygen reduction pathways under acidic conditions. We find that both 4e and 2e pathways involves the formation of an OOH intermediate, which breaks its O–OH bond in the 4e pathway but is reduced to H2O2 in the 2e pathway. We show that at 0.8 V vs. SHE, the 4e pathway is preferred at both FeN4 and Fe(CN)N4 centers, but the 2e pathway is kinetically also likely on the Fe(CN)N4 center. The O–OH bond breaking of OOH is the key kinetic step, which has a similar free energy barrier to the OH reduction on the FeN4 center, and is the rate-determining step on the Fe(CN)N4 center. Due to the high adsorption energy of Fe towards the fifth ligand, such as OH and CN, we expect that the active site of the real Fe/N/C catalyst is the five coordinated Fe center. We found that the barrier of the O–OH bond breaking step is not sensitive to potential and a Tafel slope of 60 mV is predicted for the ORR on the Fe(CN)N4 center, which is consistent with experimental observation.
Co-reporter:Sheng-Cai Zhu, Song-Hai Xie, and Zhi-Pan Liu
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 18) pp:3162-3168
Publication Date(Web):September 2, 2014
DOI:10.1021/jz5016247
Bicrystalline materials have wide applications from silicon chips to photocatalysis, but the controlled synthesis of nanocrytals with ordered phase junction has been challenging, in particular via chemical synesthetic routes. Here, we propose a general strategy to design biphase crystals formed via partial solid-to-solid phase transition with perfect phase junction, e.g., being atomically sharp and built of two particular sets of epitaxially joined planes of the two component phases, and present such an example by designing, synthesizing, and characterizing the interface of two TiO2 phases, namely, TiO2-B/anatase biphase nanocrystals that are obtained conveniently via one-pot chemical synthesis. Our design strategy classifies the common solid-to-solid phase transition into three types that are distinguishable by using the newly developed stochastic surface walking (SSW) method for unbiased pathway sampling. Only Type-I crystal is predicted to possess perfect phase junction, where the phase transition involves one and only one propagation direction featuring single pathway phase transition containing only one elementary kinetic step. The method is applicable for the understanding and the design of heterophase materials via partial phase transition in general.Keywords: heterophase crystal; orientation relation; phase junction; photocatalysis; solid-to-solid phase transition; TiO2-B/anatase;
Co-reporter:Ya-Hui Fang ; Guang-Feng Wei
The Journal of Physical Chemistry C 2014 Volume 118(Issue 7) pp:3629-3635
Publication Date(Web):January 30, 2014
DOI:10.1021/jp411531f
To understand the potential-dependent kinetics of reactions at the solid–liquid interface, we derive a constant-charge reaction theory for understanding the coupled charge transfer during the chemical bond making/breaking. The charge transfer coefficient (CTC) for reactions at the solid–liquid interface is shown to be linearly proportional to the electrochemical potential change from the initial state to the transition state as well the interface differential capacitance at the constant-charge model, and can be further related to the net dipole change normal to the surface during the reaction. Using the constant-charge theory, the CTC can be explicitly calculated on the basis of the first principles calculations without the need to assume the redox behavior of the elementary reactions and thus provide a unique possibility to evaluate and compare the magnitude of CTC for different reactions across different surfaces. By examining a series of interface reactions and comparing the calculated CTC values, we propose simple rules to understand and predict the charge transfer coefficient of three classes of the interface elementary reactions. The role of surface dipole, solvation, and molecular adsorption strength on the CTC can now be clarified from first principles calculations.
Co-reporter:Xiao-Jie Zhang, Cheng Shang, and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 7) pp:3252-3260
Publication Date(Web):May 28, 2013
DOI:10.1021/ct400238j
It is of general concern whether the automated structure prediction of unknown material without recourse to any knowledge from experiment is ever possible considering the daunting complexity of potential energy surface (PES) of material. Here we demonstrate that the stochastic surface walking (SSW) method can be a general and promising solution to this ultimate goal, which is applied to assemble carbon fullerenes containing up to 100 atoms (including 60, 70, 76, 78, 80, 84, 90, 96, and 100 atoms) from randomly distributed atoms, a long-standing challenge in global optimization. Combining the SSW method with a parallel replica exchange algorithm, we can locate the global minima (GM) of these large fullerenes efficiently without being trapped in numerous energy-nearly degenerate isomers. Detailed analyses on the SSW trajectories allow us to rationalize how and why the SSW method is able to explore the highly complex PES, which highlights the abilities of SSW method for surmounting the high barrier and the preference of SSW trajectories to the low energy pathways. The work demonstrates that the parallel SSW method is a practical tool for predicting unknown materials.
Co-reporter:Cheng Shang and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 3) pp:1838-1845
Publication Date(Web):February 5, 2013
DOI:10.1021/ct301010b
We propose an unbiased general-purpose potential energy surface (PES) searching method for both the structure and the pathway prediction of a complex system. The method is based on the idea of bias-potential-driven dynamics and Metropolis Monte Carlo. A central feature of the method is able to perturb smoothly a structural configuration toward a new configuration and simultaneously has the ability to surmount the high barrier in the path. We apply the method for locating the global minimum (GM) of short-ranged Morse clusters up to 103 atoms starting from a random structure without using extra information from the system. In addition to GM searching, the method can identify the pathways for chemical reactions with large dimensionality, as demonstrated in a nanohelix transformation containing 222 degrees of freedoms.
Co-reporter:Xiao-Jie Zhang, Cheng Shang, and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 12) pp:5745-5753
Publication Date(Web):November 14, 2013
DOI:10.1021/ct4008475
Toward the activity prediction with large-scale computations, here a double-ended surface walking (DESW) method is developed for connecting two minima on a potential energy surface (PES) and locating the associated transition state (TS) using only the first derivatives. The method operates two images starting from the initial and the final states, respectively, to walk in a stepwise manner toward each other. The surface walking involves repeated bias potential addition and local relaxation with the constrained Broyden dimer method to correct the walking direction. We apply the method to a model PES, a large set of gas phase Baker reactions, and complex surface catalytic reactions, which demonstrates that the DESW method can establish a low energy pathway linking two minima even without iterative optimization of the pathway, from which the TS can be located readily. By comparing the efficiency of the new method with the existing methods, we show that the DESW method is much less computationally demanding and is applicable for reactions with complex PESs. We hope that the DESW method may be integrated with the PES sampling methods for automated reaction prediction.
Co-reporter:Guang-Feng Wei and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 42) pp:18555-18561
Publication Date(Web):11 Sep 2013
DOI:10.1039/C3CP53758G
The electrocatalytic oxygen reduction reaction (ORR) on nanoparticles has attracted much attention in recent years for its significance in fuel cell applications. Here by combining density functional theory (DFT) calculations with the periodic continuum solvation model based on modified-Poisson–Boltzmann (CM-MPB) electrostatics, we analyzed the ORR activity on a set of differently sized Pt nanoparticles in order to identify the optimum particles for a better designed catalyst. We show that Pt nanoparticles of ∼2 nm size have the highest ORR mass activity, which is attributed to the variation of the effective reaction sites on the exposed {111} facet at the electrochemical conditions. We propose a type of a new nanocatalyst for the electrocatalytic oxygen reduction based on the knowledge from large-scale first principles simulations on Pt nanoparticles. The new catalyst has inert metal Au as the frame for the Pt nanoparticle and exposed Pt{111} sites are the active site for oxygen reduction. Such an architecture can not only prevent the initial O corrosion at the edge sites but also significantly improve the activity. The theoretical work provides a promising new direction for the rational design of a stable and active ORR catalyst via nano-structure engineering.
Co-reporter:Ye-Fei Li and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 4) pp:1082-1087
Publication Date(Web):22 Nov 2012
DOI:10.1039/C2CP44137C
This work explores thoroughly the reaction network of the partial oxidation of phenylmethanol at the TiO2–solvent interface under photocatalytic conditions by using a first-principles continuum solvation method. We demonstrate that the photocatalytic oxidation of phenylmethanol has a complex reaction network with dual pathways. The dimer pathway dominates the mechanism under aerobic conditions and a [C6H5CH(OH)O]2 peroxo dimer is the key intermediate, the decomposition of which leads to an unusual O exchange phenomenon.
Co-reporter:Ya-Hui Fang, Guang-Feng Wei, and Zhi-Pan Liu
The Journal of Physical Chemistry C 2013 Volume 117(Issue 15) pp:7669-7680
Publication Date(Web):March 25, 2013
DOI:10.1021/jp400608p
Hydrogen evolution reaction (HER: H+ + e– → 1/2H2) on metals exhibits the characteristic kinetics of electrocatalytic process. Here a theoretical method based on the constant-charge first principles periodic continuum solvation model is proposed to resolve the potential-dependent reaction kinetics on Pt and Au surfaces, and the quantitative linkage is established between the Tafel kinetics (current vs potential) and the electrochemical condition, including the surface structure, the surface charging, and the coverage. The theoretical Tafel slopes for HER are determined to be 83 mV on Pt(111) and 70 mV on Pt(100), which are generally associated with the reactions involving the minority weakly adsorbed H, i.e. the atop H above 1 ML on Pt(111) and the bridging H above 1.5 ML on Pt(100). The mechanism and the contribution of each pathway (Volmer, Tafel, and Heyrovsky pathways) are determined quantitatively. It is revealed that HER at the minority surface steps has a much higher activity than at terraces, which is responsible for the overall activity on a typical Pt electrode. The theoretical model here paved the way toward the large-scale computational screening for both active and economic hydrogen electrode.
Co-reporter:Shao-Xiong Luo, Lu Wei, Xin-Hai Zhang, Min Hwee Lim, K. X. Vivian Lin, M. H. Valerie Yeo, Wen-Hua Zhang, Zhi-Pan Liu, David J. Young, and T. S. Andy Hor
Organometallics 2013 Volume 32(Issue 10) pp:2908-2917
Publication Date(Web):May 13, 2013
DOI:10.1021/om400028n
Ir(III) complexes of cyclometalating ligands derived from the natural product cinchonine and bent (4,6-bis(diphenylphosphino)phenoxazine (Nixantphos), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos)) and planar diphosphine ligands (1,2-bis(diphenylphosphino)benzene (dppb)) exhibit good luminescence with quantum efficiencies higher than those of their parent congeners. Steric hindrance by both the bulky cinchonine-derived ligand and bent diphosphine could limit nonradiative energy transfer. The cinchonine-derived and parent complexes cover a broad emission range from 472 to 569 nm with quantum efficiencies up to 0.38 and lifetimes from 0.01 to 0.46 μs in degassed CH2Cl2 solution at room temperature. DFT calculations on selected examples are in good agreement with solid-state structures determined crystallographically and accurately predict wavelengths of emission by excited electron decay from a quinoline-centered orbital to an Ir 5d–phenyl molecular orbital. The complex [(pcn)2Ir(Nixantphos)][PF6] (2; pcn = 2′-phenyl-9-O-benzyl-10,11-dihydrocinchonine-C2,N) exhibits the highest quantum yield and could detect electron-deficient aromatic species at ppm levels.
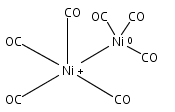
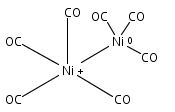
Co-reporter:Jieming Cui, Xiaojie Zhou, Guanjun Wang, Chaoxian Chi, Zhipan Liu, and Mingfei Zhou
The Journal of Physical Chemistry A 2013 Volume 117(Issue 33) pp:7810-7817
Publication Date(Web):July 29, 2013
DOI:10.1021/jp405250y
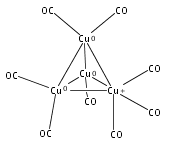
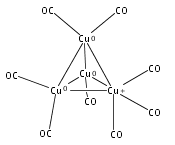
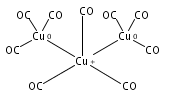
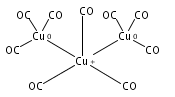
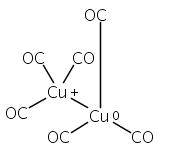
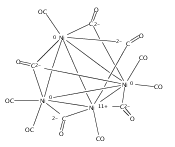
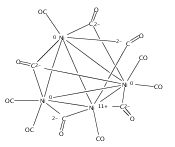
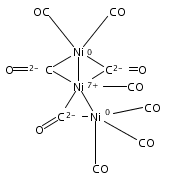
Infrared spectra of mass-selected homoleptic copper carbonyl cluster cations including dinuclear Cu2(CO)6+ and Cu2(CO)7+, trinuclear Cu3(CO)7+, Cu3(CO)8+, and Cu3(CO)9+, and tetranuclear Cu4(CO)8+ are measured via infrared photodissociation spectroscopy in the carbonyl stretching frequency region. The structures are established by comparison of the experimental spectra with simulated spectra derived from density functional calculations. The Cu2(CO)6+ cation is characterized to have an unbridged D3d structure with a Cu–Cu half bond. The Cu2(CO)7+ cation is determined to be a weakly bound complex involving a Cu2(CO)6+ core ion. The trinuclear Cu3(CO)7+ and Cu3(CO)8+ cluster cations are determined to have triangle Cu3 core structures with C2 symmetry involving two Cu(CO)3 groups and one Cu(CO)x group (x = 1 or 2). In contrast, the trinuclear Cu3(CO)9+ cluster cation is determined to have an open chain-like (OC)3Cu–Cu(CO)3–Cu(CO)3 structure. The tetranuclear Cu4(CO)8+ cluster cation is characterized to have a tetrahedral Cu4+ core structure with all carbonyl groups terminally bonded.
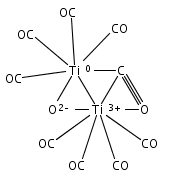
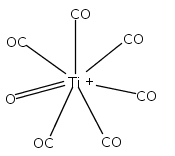
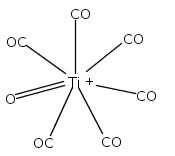
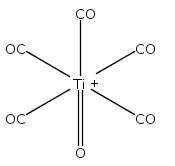
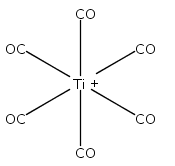
Co-reporter:Xiaojie Zhou, Jieming Cui, Zhen Hua Li, Guanjun Wang, Zhipan Liu, and Mingfei Zhou
The Journal of Physical Chemistry A 2013 Volume 117(Issue 7) pp:1514-1521
Publication Date(Web):January 18, 2013
DOI:10.1021/jp3120429
Mononuclear and dinuclear titanium carbonyl cation complexes including Ti(CO)6+, Ti(CO)7+, TiO(CO)5+, Ti2(CO)9+ and Ti2O(CO)9+ are produced via a laser vaporization supersonic cluster source. The ions are mass selected in a tandem time-of-flight mass spectrometer and studied with infrared photodissociation spectroscopy in the CO stretching frequency region. The structures are established by comparison of the experimental spectra with simulated spectra derived from density functional calculations. Only one IR band is observed for the 15-electron Ti(CO)6+ cation, which is characterized to have an octahedral Oh structure. The Ti(CO)7+ cation is determined to be a weakly bound complex involving a Ti(CO)6+ core ion instead of the seventh coordinated ion. The TiO(CO)5+ cation has a completed coordination sphere with a C4v structure. The Ti2(CO)9+ cation is determined to have a doublet Cs structure with two four-electron donor side-on bridging CO groups and one semibridging CO group. The Ti2O(CO)9+ cation has a doublet Cs structure involving a planar cyclic Ti2O(η2-μ-CO) core with a four electron donor side-on bridging CO. Bonding analysis indicates that the Ti2(CO)9+ and Ti2O(CO)9+ cations each have a Ti–Ti single bond. The results suggest that metal–metal multiple bonding is not favorable, and the oxophilic titanium centers failed to satisfy the 18-electron configuration in these metal carbonyl complexes.
Co-reporter:Cheng Shang and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 7) pp:2215-2222
Publication Date(Web):May 23, 2012
DOI:10.1021/ct300250h
To predict the chemical activity of new matter is an ultimate goal in chemistry. The identification of reaction pathways using modern quantum mechanics calculations, however, often requires a high demand in computational power and good chemical intuition on the reaction. Here, a new reaction path searching method is developed by combining our recently developed transition state (TS) location method, namely, the constrained Broyden dimer method, with a basin-filling method via bias potentials, which allows the system to walk out from the energy traps at a given reaction direction. In the new method, the reaction path searching starts from an initial state without the need for preguessing the TS-like or final state structure and can proceed iteratively to the final state by locating all related TSs and intermediates. In each elementary reaction step, a reaction direction, such as a bond breaking, needs to be specified, the information of which is refined and preserved as a normal mode through biased dimer rotation. The method is tested successfully on the Baker reaction system (50 elementary reactions) with good efficiency and stability and is also applied to the potential energy surface exploration of multistep reaction processes in the gas phase and on the surface. The new method can be applied for the computational screening of new catalytic materials with a minimum requirement of chemical intuition.
Co-reporter:Dong Chen, Ya-Hui Fang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 48) pp:16612-16617
Publication Date(Web):03 Aug 2012
DOI:10.1039/C2CP42149F
Water electrolysis is an important route to large-scale hydrogen production using renewable energy, in which the oxygen evolution reaction (OER: 2H2O → O2 + 4H+ + 4e−) causes the largest energy loss in traditional electrocatalysts involving Ru–Ir mixed oxides. Following our previous mechanistic studies on the OER on RuO2(110) (J. Am. Chem. Soc. 2010, 132, 18214), this work aims to provide further insight into the key parameters relevant to the activity of OER catalysts by investigating a group of rutile-type binary metal oxides, including RuNiO2, RuCoO2, RuRhO2, RuIrO2 and OsIrO2. Two key aspects are focused on, namely the surface O coverage at the relevant potential conditions and the kinetics of H2O activation on the O-covered surfaces. The O coverage for all the oxides investigated here is found to be 1 ML at the concerned potential (1.23 V) with all the exposed metal cations being covered by terminal O atoms. The calculated free energy barrier for the H2O dissociation on the O covered surfaces varies significantly on different surfaces. The highest OER activity occurs at RuCoO2 and RuNiO2 oxides with a predicted activity about 500 times higher than pure RuO2. On these oxides, the surface bridging O near the terminal O atom has a high activity for accepting the H during H2O splitting. It is concluded that while the differential adsorption energy of the terminal O atom influences the OER activity to the largest extent, the OER activity can still be tuned by modifying the electronic structure of surface bridging O.
Co-reporter:Jieming Cui;Xiaopeng Xing;Chaoxian Chi;Guanjun Wang;Zhipan Liu;Mingfei Zhou
Chinese Journal of Chemistry 2012 Volume 30( Issue 9) pp:2131-2137
Publication Date(Web):
DOI:10.1002/cjoc.201200595
Abstract
Infrared spectra of mass-selected homoleptic dinuclear palladium carbonyl cluster cations Pd2(CO)n+ (n=5–8) are measured via infrared photodissociation spectroscopy in the carbonyl stretching frequency region. The structures are established by comparison of the experimental spectra with simulated spectra derived from density functional calculations. The Pd2(CO)5+ cation is characterized to have two weakly semibridging CO groups with C2 symmetry. The Pd2(CO)6+ and Pd2(CO)7+ cations are determined to involve one weakly semibridging CO group. The Pd2(CO)8+ cation is a CO coordination saturated cluster, which is determined to have a D2d structure with all of the carbonyl groups terminally bonded. Bonding analysis indicates that these cluster cations each has a PdPd half bond. The PdPd distance increases with the number of CO ligands.
Co-reporter:Guang-Feng Wei, Ya-Hui Fang, and Zhi-Pan Liu
The Journal of Physical Chemistry C 2012 Volume 116(Issue 23) pp:12696-12705
Publication Date(Web):May 22, 2012
DOI:10.1021/jp3034616
Oxygen reduction is a critical reaction in the global energy cycle and a vital catalytic process in fuel cells. To date, the atomic level picture on how oxygen is electrocatalytically reduced on the traditional Pt catalyst is not established yet and the design of both active and economic catalysts remains a great challenge. Here first principles based theoretical methods can for the first time resolve the Tafel behavior and the polarization kinetics for oxygen reduction reaction (ORR) on Pt in aqueous soundings and reveal the origin of some key problems, mainly associated with the low intrinsic activity and the rapid poisoning of the electrocatalyst. The atomic level mechanism of ORR on Pt at the concerned potentials (∼0.8 V) is established, in which the critical surface coverage to achieve the reaction equilibrium is identified to be 0.25 ML O coverage. From the computed Tafel curve, the proton-coupled O–O bond breaking, i.e. H+ + e + O2ad → O + OH, is assigned to be the major O2 reduction channel on Pt; and the reaction is quenched at the high potentials due to the presence of the surface O/OH/H2O network that prevents the adsorption of bidentate O2. We predict that a qualified ORR catalyst must allow bidentate O2 adsorption under the equilibrium between adsorbed O and H2O in solution at the concerned potential.
Co-reporter:Guang-Feng Wei and Zhi-Pan Liu
Energy & Environmental Science 2011 vol. 4(Issue 4) pp:1268-1272
Publication Date(Web):25 Feb 2011
DOI:10.1039/C0EE00762E
Pt metal, when used as a cathode for oxygen reduction (O2 + 4H+ + 4e− → 2H2O), suffers from high overpotential and catalyst corrosion. Here, first-principles based theoretical methods for electrochemical systems are utilized to identify the critical factors affecting cathode performance. By analyzing a large set of Pt alloys, we show that alloys are in general less stable than Pt at the same O coverage under electrochemical conditions, and that maintaining a zero O coverage at the working potentials (e.g. 0.9 V) is key to achieve both high activity and stability. Two quantities, i.e. the surface corrosion energy and the free energy barrier to OOH dissociation, are found to be the main descriptors for the stability and activity. A Pt2Mo skin alloy is discovered to be a good candidate for an oxygen reduction cathode. The theoretical framework provides a new route for the rational design of oxygen reduction catalysts.
Co-reporter:Cheng Shang
Journal of the American Chemical Society 2011 Volume 133(Issue 25) pp:9938-9947
Publication Date(Web):May 24, 2011
DOI:10.1021/ja203468v
Whether gold is catalytically active on its own has been hotly debated since the discovery of gold-based catalysis in the 1980s. One of the central controversies is on the O2 activation mechanism. This work, by investigating aerobic phenylethanol oxidation on gold nanoparticles in aqueous solution, demonstrates that gold nanoparticles are capable to activate O2 at the solid–liquid interface. Extensive density functional theory (DFT) calculations combined with the periodic continuum solvation model have been utilized to provide a complete reaction network of aerobic alcohol oxidation. We show that the adsorption of O2 is very sensitive to the environment: the presence of water can double the O2 adsorption energy to ∼0.4 eV at commonly available edge sites of nanoparticles (∼4 nm) because of its strongly polarized nature in adsorption. In alcohol oxidation, the hydroxyl bond of alcohol can break only with the help of an external base at ambient conditions, while the consequent α-C—H bond breaking occurs on pure Au, both on edges and terraces, with a reaction barrier of 0.7 eV, which is the rate-determining step. The surface H from the α-C—H bond cleavage can be easily removed by O2 and OOH via a H2O2 pathway without involving atomic O. We find that Au particles become negatively charged at the steady state because of a facile proton-shift equilibrium on surface, OOH + OH ↔ O2 + H2O. The theoretical results are utilized to rationalize experimental findings and provide a firm basis for utilizing nanoparticle gold as aerobic oxidation catalysts in aqueous surroundings.
Co-reporter:Ye-Fei Li
Journal of the American Chemical Society 2011 Volume 133(Issue 39) pp:15743-15752
Publication Date(Web):August 31, 2011
DOI:10.1021/ja206153v
TiO2 nanoparticles have been widely utilized in photocatalysis, but the atomic level understanding on their working mechanism falls much short of expectations. In particular, the correlation between the particle structure and the photocatalytic activity is not established yet, although it was observed that the activity is sensitive to the particle size and shape. This work, by investigating a series of TiO2 anatase nanoparticles with different size and shape as the photocatalyst for water oxidation, correlates quantitatively the particle size and shape with the photocatalytic activity of the oxygen evolution reaction (OER). Extensive density functional theory (DFT) calculations combined with the periodic continuum solvation model have been utilized to compute the electronic structure of nanoparticles in aqueous solution and provide the reaction energetics for the key elementary reaction. We demonstrate that the equilibrium shape of nanoparticle is sensitive to its size from 1 to 30 nm, and the sharp crystals possess much higher activity than the flat crystals in OER, which in combination lead to the morphology dependence of photocatalytic activity. The conventionally regarded quantum size effect is excluded as the major cause. The physical origin for the shape–activity relationship is identified to be the unique spatial separation/localization of the frontier orbitals in the sharp nanoparticles, which benefits the adsorption of the key reaction intermediate (i.e., OH) in OER on the exposed five-coordinated Ti of {101} facet. The theoretical results here provide a firm basis for maximizing photocatalytic activity via nanostructure engineering and are also of significance for understanding photocatalysis on nanomaterials in general.
Co-reporter:Qiuyang Sun;Zhipan Liu
Frontiers of Chemistry in China 2011 Volume 6( Issue 3) pp:
Publication Date(Web):2011 September
DOI:10.1007/s11458-011-0250-9
The efficient fixation and utilization of CO2 has been consistently pursued by chemists for decades. Although Cu-based catalysts, e.g., Cu/ZnO/Al2O3, have been widely used in industry for methanol synthesis from CO2 hydrogenation (CO2 + 3H2→H3COH + H2O), many issues on the mechanism and the kinetics remain largely uncertain. For example, the surface site for CO2 activation and the synergetic effect between Cu and oxide have been hotly debated in literature. In the past few years, theoretical modeling on pure Cu surfaces and Cu/oxide interfaces has been utilized to provide insight into these important questions. Here we will review the recent theoretical advances on simulating this complex heterogeneous catalytic process with first principles density functional theory (DFT) calculations and kinetics modeling. The theoretical results on the mechanism and the kinetics are compared and summarized.
Co-reporter:Ya-Hui Fang
The Journal of Physical Chemistry C 2011 Volume 115(Issue 35) pp:17508-17515
Publication Date(Web):August 3, 2011
DOI:10.1021/jp205376b
To catalyze oxygen reduction reaction (ORR) under electrochemical conditions approaching its thermodynamic limit, i.e., 1.23 V vs NHE, has been consistently pursued by chemists. It is known that metal electrodes, even noble metals, undergo severe corrosion at the high potential, accompanied with the rapid decrease in activity. A comprehensive understanding of both the stability and the catalytic activity of the Pt surface is urgently called for toward the rational design of a new ORR catalyst. This work, by utilizing chemically inert Au as a modifier to the Pt surface, investigated the stability and the activity of a set of Au/Pt composites using first-principles-based theoretical methods designed for the modeling of solid/liquid electrocatalysis. By computing the surface phase diagram and the corrosion thermodynamics, we demonstrate that the presence of Au can remarkably reduce the in situ O atom coverage to be below a critical local 0.5 monolayer (ML) and thus protect the neighboring Pt sites from corrosion. The AuPt surface alloy with a very low amount of Au dispersing in the Pt surface layer is sufficient to stabilize the whole catalyst surface. With the calculated ORR profiles, we show that ORR activity in AuPt surface alloys is not sensitive to the Au concentration, and a good activity is maintained with the Au concentration up to 7/8 ML. Fundamentally, this is because the minimum active site of ORR requires only two neighboring Pt atoms (with an OOH pathway), and the majority of surface Pt sites on pure Pt are in fact nonactive spectators that are terminated by O atoms at the working potentials. The key factors controlling the ORR activity and surface stability are therefore unified as the minimum O coverage and the minimum exposure of Pt (active) sites. The theory presented here suggests that the structural engineering to separate active sites (e.g., two Pt atoms) by inert elements (e.g., Au) is an effective approach for yielding a stable, active, and economic catalyst. Experimental observations on Au/Pt composite catalysts are discussed in the context of current findings, focusing on the thermodynamic tendency for AuPt surface alloy formation.
Co-reporter:Ya-Hui Fang
Journal of the American Chemical Society 2010 Volume 132(Issue 51) pp:18214-18222
Publication Date(Web):December 6, 2010
DOI:10.1021/ja1069272
How to efficiently oxidize H2O to O2 (H2O → 1/2O2 + 2H+ + 2e−) is a great challenge for electrochemical/photo water splitting owing to the high overpotential and catalyst corrosion. Here extensive periodic first-principles calculations integrated with modified-Poisson−Boltzmann electrostatics are utilized to reveal the physical origin of the high overpotential of the electrocatalytic oxygen evolution reaction (OER) on RuO2(110). By determining the surface phase diagram, exploring the possible reaction channels, and computing the Tafel lines, we are able to elucidate some long-standing puzzles on the OER kinetics from the atomic level. We show that OER occurs directly on an O-terminated surface phase above 1.58 V vs NHE, but indirectly on a OH/O mixed phase below 1.58 V by converting first the OH/O mixed phase to the O-terminated phase locally. The rate-determining step of OER involves an unusual water oxidation reaction following a Eley−Rideal-like mechanism, where a water molecule from solution breaks its OH bond over surface Os with concurrent new O—OH bond formation. The free energy barrier is 0.74 eV at 1.58 V, and it decreases linearly with the increase of potential above 1.58 V (a slope of 0.56). In contrast, the traditionally regarded surface oxygen coupling reaction with a Langmuir−Hinshelwood mechanism is energetically less favored and its barrier is weakly affected by the potential. Fundamentally, we show that the empirical linear barrier∼potential relation is caused by the linear structural response of the solvated transition state to the change of potential. Finally, the general strategy for finding better OER anode is also presented.
Co-reporter:Ye-Fei Li ; Zhi-Pan Liu ; LuLu Liu ;Weiguo Gao
Journal of the American Chemical Society 2010 Volume 132(Issue 37) pp:13008-13015
Publication Date(Web):August 25, 2010
DOI:10.1021/ja105340b
Due to its high overpotential and low efficiency, the conversion of water to O2 using solar energy remains a bottleneck for photocatalytic water splitting. Here the microscopic mechanisms of the oxygen evolution reaction (OER) on differently structured anatase surfaces in aqueous surroundings, namely, (101), (001), and (102), are determined and compared systematically by combining first-principles density functional theory calculations and a parallel periodic continuum solvation model. We show that OER involves the sequential removal of protons from surface oxidative species, forming surface peroxo and superoxo intermediates. The initiating step, the first proton removal, dictates the high overpotential. Only at an overpotential of 0.7 V (1.93 V vs SHE) does this rate-controlling step become surmountable at room temperature: the free energy change of the step is 0.69, 0.63, and 0.61 eV for (101), (102), and (001) surfaces, respectively. We therefore conclude that (i) OER is not sensitive to the local surface structure of anatase and (ii) visible light (<∼590 nm) is, in principle, capable of driving the photocatatlytic OER on anatase kinetically. By co-doping high-valent elements into the anatase subsurface, we demonstrate that the high overpotential of the OER can be significantly reduced, with extra occupied levels above the valence band.
Co-reporter:Cheng Shang and Zhi-Pan Liu
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 4) pp:1136-1144
Publication Date(Web):March 10, 2010
DOI:10.1021/ct9005147
To determine transition state (TS) and, thus, to predict chemical activity has been a challenging topic in theoretical simulations of chemical reactions. In particular, with the difficulty to compute the second derivative of energy (Hessian) in modern quantum mechanics packages with a non-Gaussian basis set, the location usually involves a high demand in computational power and lacks stability in the algorithm, especially for complex reaction systems with many degrees of freedom. Here, an efficient TS searching method is developed by combining the constrained Broyden minimization algorithm with the dimer method that was first proposed by Henkelman and Jónsson. In the new method, the rotation of the dimer needs only one energy and gradient calculation for determining a rotation angle; the translation of the dimer is continually carried out until a termination criterion is met, and the translational force parallel to the dimer direction is damped to optimize the searching trajectory. Based on our results of the Baker reaction system and of a heterogeneous catalytic reaction, our method is shown to increase the efficiency significantly and is also more stable in finding TSs.
Co-reporter:Qi-Jun Hong, Zhi-Pan Liu
Surface Science 2010 Volume 604(21–22) pp:1869-1876
Publication Date(Web):October 2010
DOI:10.1016/j.susc.2010.07.018
It has been a goal consistently pursued by chemists to understand and control the catalytic process over composite materials. In order to provide deeper insight on complex interfacial catalysis at the experimental conditions, we performed an extensive analysis on CO2 hydrogenation over a Cu/ZrO2 model catalyst by employing density functional theory (DFT) calculations and kinetic Monte Carlo (kMC) simulations based on the continuous stirred tank model. The free energy profiles are determined for the reaction at the oxygen-rich Cu/m-ZrO2 (2̅12) interface, where all interfacial Zr are six-coordinated since the interface accumulates oxidative species at the reaction conditions. We show that not only methanol but also CO are produced through the formate pathway dominantly, whilst the reverse-water-gas-shift (RWGS) channel has only a minor contribution. H2CO is a key intermediate species in the reaction pathway, the hydrogenation of which dictates the high temperature of CO2 hydrogenation. The kinetics simulation shows that the CO2 conversion is 1.20%, the selectivity towards methanol is 68% at 500 K and the activation energies for methanol and CO formation are 0.79 and 1.79 eV, respectively. The secondary reactions due to the product readsorption lower the overall turnover frequency (TOF) but increase the selectivity towards methanol by 16%. We also show that kMC is a more reliable tool for simulating heterogeneous catalytic processes compared to the microkinetics approach.
Co-reporter:Ya-Hui Fang and Zhi-Pan Liu
The Journal of Physical Chemistry C 2010 Volume 114(Issue 9) pp:4057-4062
Publication Date(Web):February 17, 2010
DOI:10.1021/jp9111734
As an important concern in both science and industry, metal corrosion at solid/liquid interfaces is not well understood at the atomic level. The challenge to investigators lies in the simultaneous consideration of the extended solid surface, the electrochemical potential, and the water solution. The work presented here represents the first theoretical attempt to elucidate the oxidation mechanism of the Pt electrode under electrochemical conditions by exploring the oxidation kinetics of differently structured Pt surfaces, including Pt(111), Pt(211), and Pt(100). We show that the most abundant and close-packed (111) surface in Pt metal can be oxidized because of the presence of surface OH. The corrosion is self-accelerated kinetically once the defects are created, as demonstrated by the low kinetic barrier of oxidation on Pt(211). By contrast, the open Pt(100) facet is very inert toward surface oxidation. Apart from the revealed surface-structure sensitivity, Pt corrosion is also strongly affected by the local oxygen coverage as pinned by the electrochemical potential. For Pt(111), the subsurface oxygen formation occurs only above 0.5 ML oxygen coverage around 1.1 V. The kinetics model for the surface oxidation proposed in this work provides new insights for designing the next generation of anticorrosion electrode materials in fuel cells.
Co-reporter:Qian-Lin Tang and Zhi-Pan Liu
The Journal of Physical Chemistry C 2010 Volume 114(Issue 18) pp:8423-8430
Publication Date(Web):April 2, 2010
DOI:10.1021/jp100864j
The water−gas shift (WGS) reaction (H2O + CO → H2 + CO2) is regarded as a key catalytic process in a future hydrogen economy. In this report, first-principles density functional theory (DFT) calculations have been utilized to identify the WGS mechanism over a Cu/oxide model catalyst, Cu/ZrO2. The catalytic reaction is found to occur at the Cu sites that are in the vicinity of Cu/oxides interfaces, where the Cu electronic structure is markedly modified by the oxygen-rich Cu/oxides interface. DFT-based microkinetic modeling further shows that a COOH-involved mechanism is responsible for the WGS reaction, with the H2O dissociation step being rate-controlling. By comparing the reaction thermodynamics and kinetics over three systems, namely, Cu/ZrO2, unsupported Cu strip, and Cu(111), we demonstrate that positively charged Cu clusters afford much enhanced catalytic activity in H2O dissociation. The ZrO2 support acts as a charge buffer to accept/release electrons from/to the Cu particle. The oxygen-rich metal-oxide interface, although not directly involved in catalysis, acts as a key promoter in enhancing catalytic activity in Cu-based catalysis.
Co-reporter:Bin Peng, Hui-Fang Wang, Zhi-Pan Liu and Wen-Bin Cai
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:3102-3107
Publication Date(Web):February 1, 2010
DOI:10.1021/jp910497n
In situ electrochemical surface-enhanced infrared absorption spectroscopy (EC-SEIRAS) together with a periodic density functional theory (DFT) calculation has been initially applied to investigate the mechanism of formic acid electro-oxidation on Sb-modified Pt (Sb/Pt) electrode. EC-SEIRAS measurement reveals that the main formic acid oxidation current on Sb/Pt electrode is ca. 10-fold enhanced as compared to that on clean Pt electrode, mirrored by nearly synchronous decrease of the CO and formate surface species, suggesting a “non-formate” oxidation as the main pathway on the Sb/Pt electrode. On the basis of the calculations from periodic DFT, the catalytic role of Sb adatoms can be rationalized as a promoter for the adsorption of the CH-down configuration but an inhibitor for the adsorption of the O-down configuration of formic acid, kinetically facilitating the complete oxidation of HCOOH into CO2. In addition, Sb modification lowers the CO adsorption energy on Pt, helps to mitigate the CO poisoning effect on Pt.
Co-reporter:YaHui Fang;ZhiPan Liu
Science China Chemistry 2010 Volume 53( Issue 3) pp:543-552
Publication Date(Web):2010 March
DOI:10.1007/s11426-010-0047-6
Theoretical simulations on complex electrochemical processes have been developed on the basis of the understanding in electrochemistry, which has benefited from quantum mechanics calculations. This article reviews the recent progress on the theory and applications in electrocatalysis. Two representative reactions, namely water electrolysis and oxygen reduction, are selected to illustrate how the theoretical methods are applied to electrocatalytic reactions. The microscopic nature of these electrochemical reactions under the applied potentials is described and the understanding of the reactions is summarized. The thermodynamics and kinetics of the electrochemical reactions affected by the interplay of the electrochemical potential, the bonding strength and the local surface structure are addressed at the atomic level.
Co-reporter:Cheng Shang
The Journal of Physical Chemistry C 2010 Volume 114(Issue 40) pp:16989-16995
Publication Date(Web):June 22, 2010
DOI:10.1021/jp102477g
Although a poor catalyst under dry conditions, γ-Al2O3-supported gold (Au/γ-Al2O3) turns out to be a superior CO oxidation catalyst under moisture conditions. In this work, extensive density functional theory calculations have been carried out to investigate the physical origin of the moisture promotion effect. By supporting Au strips on the two most stable γ-Al2O3 surfaces, namely, the (110) and (100) faces, we show that the majority (110) surface is catalytically inert due to the saturation of Al cationic sites by dissociated H2O. On the other hand, the minority (100) surface in combination with Au is responsible for CO oxidation activity, where O2 can adsorb at the Au/γ-Al2O3(100) interface with a tilted Au−O−O−Al5c configuration. In the presence of coadsorbed H2O and CO, the adsorption energy of O2 reaches to 0.7 eV. We find that H2O enables the direct dissociation of the reaction intermediate cis-OCOO produced by the bimolecular coupling between CO and O2, whereas an extra cis-to-trans rotation of OCOO is required in the absence of H2O. In the H2O-assisted pathway, no atomic oxygen is produced, and the overall barrier is only 0.25 eV, which is 0.28 eV lower than that without H2O. By electronic structure analyses, we suggest that a modest acidity of the γ-Al2O3 (100) surface contributes to the O2 adsorption, although Al2O3 lacks the d-states that were shown to be important for O2 activation.
Co-reporter:Ya-Hui Fang and Zhi-Pan Liu
The Journal of Physical Chemistry C 2009 Volume 113(Issue 22) pp:9765-9772
Publication Date(Web):May 8, 2009
DOI:10.1021/jp901091a
Electrochemical reactions catalyzed by metal electrode, despite their huge importance in industry, are not well understood at the atomic level. In relevance to water electrolysis, the oxygen coupling reaction on Pt metal surfaces is systematically investigated in this work by combining periodic density functional theory calculations with a new theoretical approach to mimic the electrochemical environment. In our approach, the surface is explicitly polarized by adding/subtracting charges and the counter charges are placed as Gaussian-distributed plane charges in a vacuum. With this method, the surface phase diagrams for both the closed-packed Pt(111) and stepped Pt(211) are determined, which demonstrates that stepped surface sites can better accumulate oxidative species and thus reach to a higher local O coverage compared to Pt(111) at a given potential. The water environment is proved to affect the phase diagram marginally. By fully exploring the possible oxygen coupling channels on Pt surfaces, we show that the oxygen coupling reaction is kinetically difficult on metallic Pt surfaces below 1.4 V. There is no facile O coupling channels on Pt(111), as the barriers are no less than 1 eV. Although an O + OH → OOH reaction can eventually occur at the stepped sites with an increase of local O coverage and the calculated barrier is lower than 0.7 eV at 1.4 V (NHE), at such high potentials the (111) surface can already undergo surface oxidation due to the penetration of oxygen into sublayers. The theory thus indicates that oxygen evolution on Pt anode occurs on Pt surface oxides as dictated by thermodynamics and also demonstrates that the local surface structure and coverage can be more important in affecting the barrier of surface reactions than the electric fields.
Co-reporter:Hui-Fang Wang and Zhi-Pan Liu
The Journal of Physical Chemistry C 2009 Volume 113(Issue 40) pp:17502-17508
Publication Date(Web):September 11, 2009
DOI:10.1021/jp9059888
As an important class of catalytic reactions, the reaction at solid/liquid interfaces is less understood at the atomic level. From a theoretical point of view, the difficulty lies at the simultaneous consideration of the extended solid surface and the dynamic liquid environment. In taking the oxidation of formic acid (HCOOH → CO2 + H2) at the Pt(111)/H2O interface as the model system that has great potentials in direct fuel cells applications, this work combines density functional theory (DFT) slab calculations with a continuum solvation model to simulate the reactions at the metal/H2O interface for the first time. The solvation effect is treated by including (i) a few explicit water molecules as the core solvation shell and (ii) an implicit continuum solvation model to take into account the long-range electrostatic interaction from water solution. We show that formic acid can be directly oxidized to CO2 only in the presence of preadsorbed formate. Although the formate itself can not be oxidized to CO2 at mild conditions, it helps to stabilize the formic acid adsorption configuration with the CH bond in contact with the Pt surface, which is the precursor leading to CO2. Without the preadsorbed formate, formic acid is only able to adsorb with its carboxyl O linking to Pt, which is however difficult to decompose further. By electronic structure analyses, we show that a hydrophobic zone formed nearby the preadsorbed fomate on Pt(111) is the origin for the promoting role of formate, which demonstrates that catalytic reactions at solid/water interface can be significantly affected by modifying the affinity between surface and water.
Co-reporter:Qiang Zhao, Lei Li, Fuyou Li, Mengxiao Yu, Zhipan Liu, Tao Yi and Chunhui Huang
Chemical Communications 2008 (Issue 6) pp:685-687
Publication Date(Web):30 Nov 2007
DOI:10.1039/B712416C
A novel aggregation-induced phosphorescent emission (AIPE) was observed for iridium(III) complexes. This interesting phenomenon was attributed to the intermolecular packing, resulting in a switch from the non-emissive 3LX excited state to the emissive 3MLLCT transition, which is confirmed by X-ray diffraction studies as well as theoretical calculations.
Co-reporter:Zhi-Pan Liu ;Chuan-Ming Wang;Kang-Nian Fan
Angewandte Chemie 2006 Volume 118(Issue 41) pp:
Publication Date(Web):25 SEP 2006
DOI:10.1002/ange.200601853
Der aktiven Spezies auf der Spur: Rechnungen erklären die Bildung und Aktivität einzelner Goldatome in der heterogenkatalysierten Hydrierung von 1,3-Butadien. Die aktive Spezies ist ein AuI-Komplex, der in situ durch Reduktion von AuIII entsteht und durch ZrO2-Defekte stabilisiert wird. Die hohe katalytische Aktivität und Selektivität folgen aus der stabilen, aber flexiblen Struktur um das AuI-Zentrum, das H2 heterolytisch spaltet.
Co-reporter:Zhi-Pan Liu ;Chuan-Ming Wang;Kang-Nian Fan
Angewandte Chemie International Edition 2006 Volume 45(Issue 41) pp:
Publication Date(Web):25 SEP 2006
DOI:10.1002/anie.200601853
The origin of the species: The first principles method reveals the origin and activity of a heterogeneous Au monomer in the catalysis of a selective 1,3-butadiene hydrogenation. The active Au species is an AuI complex, produced from AuIII in situ by reduction, stabilized by ZrO2 defects. The high catalytic activity and selectivity comes from the stable, but flexible, structure of the AuI catalytic center, which dissociates H2 heterolytically.
Co-reporter:Chuan-Ming Wang, Yang-Dong Wang, Hong-Xing Liu, Zai-Ku Xie, Zhi-Pan Liu
Journal of Catalysis (4 May 2010) Volume 271(Issue 2) pp:386-391
Publication Date(Web):4 May 2010
DOI:10.1016/j.jcat.2010.02.025
As an attractive alternative to produce light olefins, methanol-to-olefins (MTO) conversion catalyzed by zeolites or zeotype materials was recently proposed to follow a hydrocarbon pool mechanism. In this contribution, the effect of the structure of methylbenzenes (MBs) in the pore of HSAPO-34 catalyst on the MTO activity and selectivity is investigated by first-principle calculations and kinetic simulations. We demonstrate that MBs with five or six methyl groups are not more active than those with fewer methyl groups. Propene is intrinsically more favorable than ethene when the reaction is not diffusion limited based on the side-chain hydrocarbon pool mechanism. Our theoretical results are consistent with some experimental observations and can be rationalized based on the shape selectivity of key reaction intermediates and transition states in the pore of catalyst.Theoretical calculations on the methanol-to-olefins conversion in HSAPO-34 reveal that hexamethylbenzene and pentamethylbenzene are not more active than methylbenzenes with fewer methyl groups, and propene is the favored product.Download high-res image (36KB)Download full-size image
Co-reporter:Zhi-Jian Yang, Ye-Fei Li, Qing-Bin Wu, Nan Ren, Ya-Hong Zhang, Zhi-Pan Liu, Yi Tang
Journal of Catalysis (13 June 2011) Volume 280(Issue 2) pp:247-254
Publication Date(Web):13 June 2011
DOI:10.1016/j.jcat.2011.03.026
Layered niobic acids (HxK1−xNb3O8, x = 0–1) are reported as new solid acid catalysts for the selective hydration of ethylene oxide (EO). They are prepared by simply calcinating Nb2O5–K2CO3 mixture followed by an ion-exchange process in HNO3 solution of different concentrations. The highest selectivity for monoethylene glycol (MEG) is achieved over 95% with EO conversion of >99% at x of 0.7 under H2O/EO ratio of 8. Combined with the results of first-principles density functional theory calculations and Hammett indicator method, it is revealed that the suitable acid strength is more crucial for MEG selectivity than acid amount. Furthermore, a self-exfoliation of layered HNb3O8 is also found during EO hydration, which proves to be another important factor for its good catalytic performance by exposing the abundant acid sites among the Nb3O8- nanosheets. The thermal stability investigation of HNb3O8 also indicates a careful selection of characterization and application way for this layered niobic acid.Graphical abstractLayered niobic acids are reported as efficient catalysts for the selective hydration of ethylene oxide. The adjustable acidity and in situ self-exfoliation effect are crucial for the good catalytic performance.Download high-res image (80KB)Download full-size imageHighlights► Layered HxK1−xNb3O8 as efficient catalyst for EO hydration. ► Suitable acid strength is crucial for high MEG selectivity. ► The self-exfoliation process is critical for good catalytic performance. ► Theoretical calculation is a valid method to characterize acid strength.
Co-reporter:Chuan-Ming Wang, Kang-Nian Fan, Zhi-Pan Liu
Journal of Catalysis (10 September 2009) Volume 266(Issue 2) pp:343-350
Publication Date(Web):10 September 2009
DOI:10.1016/j.jcat.2009.06.023
The searching for more practical applications of single-site heterogeneous catalysis has attracted much attention recently. Here the hydrogenation of acrolein on AuOH/m-ZrO2 is extensively investigated theoretically aiming to verify whether a desired selectivity toward allyl alcohol can be achieved. Although similar hydrogenation reactions have been reported for Au/oxides catalysts in experiment, it is not clear what the active Au component is. In this work we evaluate both the stability and the activity of single Au supported on monoclinic ZrO2 surfaces from first principles. Our calculated results indicate that Au clusters are the most stable form on the flat m-ZrO2 surface, while single Au cations can be available on the stepped m-ZrO2 sites below 350 K at ambient (oxidizing) conditions. Importantly, we demonstrate that the minority species, AuOH/m-ZrO2(2¯12), exhibits the desired catalytic selectivity for the hydrogenation of acrolein. The deep hydrogenation to propyl alcohol can also be prevented kinetically on this single-site heterogeneous catalyst.First principle calculations show that single Au cations present at ZrO2 stepped sites at room temperatures can selectively hydrogenate acrolein to allyl alcohol.Download high-res image (84KB)Download full-size image
Co-reporter:Qian-Lin Tang, Qi-Jun Hong, Zhi-Pan Liu
Journal of Catalysis (1 April 2009) Volume 263(Issue 1) pp:114-122
Publication Date(Web):1 April 2009
DOI:10.1016/j.jcat.2009.01.017
The efficient fixation/utilization of CO2 has been pursued by chemists for decades. In this work, the catalytic kinetics of CO2 fixation to methanol over a binary catalyst Cu/ZrO2 is investigated by first principles kinetic Monte Carlo simulation. A Cu/ZrO2 interface model is first established and the reaction network of CO2 hydrogenation is explored. In the Cu/ZrO2 system two reaction channels to methanol are identified (i) a reverse water–gas shift reaction via CO2 decomposition to CO and (ii) the well-regarded mechanism via a formate intermediate. The theoretical selectivity is determined to be 85% for methanol and 15% for CO. The removal of the oxidative species is kinetically slow. As a result, 87% of the interface sites are covered by these oxidative species, which oxidize the interface Cu. We show that the binding strength of O atom at the interface is a critical parameter determining the activity and selectivity of the catalyst.DFT based kinetic Monte Carlo simulation of CO2 fixation to methanol over Cu/ZrO2 reveals that hydrolysis is a key step in producing methanol and that the interfacial Cu is partially oxidized during reaction.Download high-res image (126KB)Download full-size image
Co-reporter:Guang-Feng Wei and Zhi-Pan Liu
Chemical Science (2010-Present) 2015 - vol. 6(Issue 2) pp:NaN1490-1490
Publication Date(Web):2014/11/26
DOI:10.1039/C4SC02806F
The restructuring of nanoparticles at the in situ condition is a common but complex phenomenon in nanoscience. Here, we present the first systematic survey on the structure dynamics and its catalytic consequence for hydrogen evolution reaction (HER) on Pt nanoparticles, as represented by a magic number Pt44 octahedron (∼1 nm size). Using a first principles calculation based global structure search method, we stepwise follow the significant nanoparticle restructuring under HER conditions as driven by thermodynamics to expose {100} facets, and reveal the consequent large activity enhancement due to the marked increase of the concentration of the active site, being identified to be apex atoms. The enhanced kinetics is thus a “byproduct” of the thermodynamical restructuring. Based on the results, the best Pt catalyst for HER is predicted to be ultrasmall Pt particles without core atoms, a size below ∼20 atoms.
Co-reporter:Guang-Feng Wei, Cheng Shang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 3) pp:NaN2087-2087
Publication Date(Web):2014/11/19
DOI:10.1039/C4CP04145C
A supported nanoparticle is dynamic in reaction conditions, but how this dynamic behavior is influenced by the support remains elusive. Using the stochastic surface walking global optimization method, herein, we report the structure, thermodynamics and catalytic properties of Pt nanoparticles inside and outside a carbon nanotube (CNT). We reveal that confined metal nanoparticles are, surprisingly, structurally more flexible at low temperatures but less likely to melt at high temperatures. By investigating the O2 interaction with a Pt15 cluster inside and outside the CNT, we find that the low temperature structure versatility facilitates the in situ creation of favorable reaction sites, and thus maintains the catalytic activity of O2 dissociation. The decrease of the population for the liquid-like structures (largely disordered) offers the higher stability of the confined nanocatalyst. The theoretical results are consistent with experimental findings for the enhanced antioxidation ability of the confined metal nanoparticles.
Co-reporter:Dong Chen, Ya-Hui Fang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 48) pp:NaN16617-16617
Publication Date(Web):2012/08/03
DOI:10.1039/C2CP42149F
Water electrolysis is an important route to large-scale hydrogen production using renewable energy, in which the oxygen evolution reaction (OER: 2H2O → O2 + 4H+ + 4e−) causes the largest energy loss in traditional electrocatalysts involving Ru–Ir mixed oxides. Following our previous mechanistic studies on the OER on RuO2(110) (J. Am. Chem. Soc. 2010, 132, 18214), this work aims to provide further insight into the key parameters relevant to the activity of OER catalysts by investigating a group of rutile-type binary metal oxides, including RuNiO2, RuCoO2, RuRhO2, RuIrO2 and OsIrO2. Two key aspects are focused on, namely the surface O coverage at the relevant potential conditions and the kinetics of H2O activation on the O-covered surfaces. The O coverage for all the oxides investigated here is found to be 1 ML at the concerned potential (1.23 V) with all the exposed metal cations being covered by terminal O atoms. The calculated free energy barrier for the H2O dissociation on the O covered surfaces varies significantly on different surfaces. The highest OER activity occurs at RuCoO2 and RuNiO2 oxides with a predicted activity about 500 times higher than pure RuO2. On these oxides, the surface bridging O near the terminal O atom has a high activity for accepting the H during H2O splitting. It is concluded that while the differential adsorption energy of the terminal O atom influences the OER activity to the largest extent, the OER activity can still be tuned by modifying the electronic structure of surface bridging O.
Co-reporter:Guang-Feng Wei and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 42) pp:NaN18561-18561
Publication Date(Web):2013/09/11
DOI:10.1039/C3CP53758G
The electrocatalytic oxygen reduction reaction (ORR) on nanoparticles has attracted much attention in recent years for its significance in fuel cell applications. Here by combining density functional theory (DFT) calculations with the periodic continuum solvation model based on modified-Poisson–Boltzmann (CM-MPB) electrostatics, we analyzed the ORR activity on a set of differently sized Pt nanoparticles in order to identify the optimum particles for a better designed catalyst. We show that Pt nanoparticles of ∼2 nm size have the highest ORR mass activity, which is attributed to the variation of the effective reaction sites on the exposed {111} facet at the electrochemical conditions. We propose a type of a new nanocatalyst for the electrocatalytic oxygen reduction based on the knowledge from large-scale first principles simulations on Pt nanoparticles. The new catalyst has inert metal Au as the frame for the Pt nanoparticle and exposed Pt{111} sites are the active site for oxygen reduction. Such an architecture can not only prevent the initial O corrosion at the edge sites but also significantly improve the activity. The theoretical work provides a promising new direction for the rational design of a stable and active ORR catalyst via nano-structure engineering.
Co-reporter:Ye-Fei Li and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 4) pp:NaN1087-1087
Publication Date(Web):2012/11/22
DOI:10.1039/C2CP44137C
This work explores thoroughly the reaction network of the partial oxidation of phenylmethanol at the TiO2–solvent interface under photocatalytic conditions by using a first-principles continuum solvation method. We demonstrate that the photocatalytic oxidation of phenylmethanol has a complex reaction network with dual pathways. The dimer pathway dominates the mechanism under aerobic conditions and a [C6H5CH(OH)O]2 peroxo dimer is the key intermediate, the decomposition of which leads to an unusual O exchange phenomenon.
Co-reporter:Cheng Shang, Xiao-Jie Zhang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 33) pp:NaN17856-17856
Publication Date(Web):2014/06/12
DOI:10.1039/C4CP01485E
The determination of crystal structures and the solid-to-solid phase transition mechanisms are two important and related subjects in material science. Here we develop an unbiased general-purpose potential energy surface (PES) searching method, namely, SSW-crystal method, for prediction of both the crystal structure and the crystal phase transition pathway. The SSW-crystal method features with stochastic surface walking (SSW) via repeated small structural perturbation by taking into account the second derivative information on both the lattice and the atom degrees of freedom. The SSW-crystal method is capable of overcoming the high barrier of phase transition and identifying the desirable phase transition reaction coordinates. By applying the SSW-crystal method to a set of examples, including SiO2 crystal up to 162 atoms per cell, Lennard-Jones model crystals up to 256 atoms, ternary SrTiO3 crystal of 50 atoms and the rutile-to-anatase TiO2 phase transition, we show that the SSW-crystal method can efficiently locate the global minimum (GM) from random initial structures without a priori knowledge of the system, and also allows for exhaustive sampling of the phase transition pathways, from which the lowest energy pathway can be obtained.
Co-reporter:Jing Sun, Ya-Hui Fang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 27) pp:NaN13740-13740
Publication Date(Web):2014/02/27
DOI:10.1039/C4CP00037D
The Fe/N/C catalysts have emerged recently as a representative class of non-Pt catalysts for oxygen electrocatalytic reduction, which could have a competitive catalytic performance to Pt. However, the nature of the catalyst remains elusive, especially on the active site structure and the electrocatalytic kinetics. Here we examine two kinds of Fe/N active sites for Fe/N/C catalysts, namely, the four-coordinated FeN4 and the five-coordinated Fe(CN)N4 centers embedded in graphene layers. By using large-scale first principles calculations with a periodic continuum solvation model based on the Modified-Poisson–Boltzmann equation (CM-MPB), we identified the four (4e) and two electron (2e) oxygen reduction pathways under acidic conditions. We find that both 4e and 2e pathways involves the formation of an OOH intermediate, which breaks its O–OH bond in the 4e pathway but is reduced to H2O2 in the 2e pathway. We show that at 0.8 V vs. SHE, the 4e pathway is preferred at both FeN4 and Fe(CN)N4 centers, but the 2e pathway is kinetically also likely on the Fe(CN)N4 center. The O–OH bond breaking of OOH is the key kinetic step, which has a similar free energy barrier to the OH reduction on the FeN4 center, and is the rate-determining step on the Fe(CN)N4 center. Due to the high adsorption energy of Fe towards the fifth ligand, such as OH and CN, we expect that the active site of the real Fe/N/C catalyst is the five coordinated Fe center. We found that the barrier of the O–OH bond breaking step is not sensitive to potential and a Tafel slope of 60 mV is predicted for the ORR on the Fe(CN)N4 center, which is consistent with experimental observation.
Co-reporter:Sheng-Cai Zhu, Shu-Hui Guan and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 27) pp:NaN18574-18574
Publication Date(Web):2016/06/20
DOI:10.1039/C6CP03673B
Solid-to-solid phase transition, although widely exploited in making new materials, challenges persistently our current theory for predicting its complex kinetics and rich microstructures in transition. The Ga2O3 α–β phase transformation represents such a common but complex reaction with marked change in cation coordination and crystal density, which was known to yield either amorphous or crystalline products under different synthetic conditions. Here we, via recently developed stochastic surface walking (SSW) method, resolve for the first time the atomistic mechanism of Ga2O3 α–β phase transformation, the pathway of which turns out to be the first reaction pathway ever determined for a new type of diffusionless solid phase transition, namely, pseudomartensitic phase transition. We demonstrate that the sensitivity of product crystallinity is caused by its multi-step, multi-type reaction pathway, which bypasses seven intermediate phases and involves all types of elementary solid phase transition steps, i.e. the shearing of O layers (martensitic type), the local diffusion of Ga atoms (reconstructive type) and the significant lattice dilation (dilation type). While the migration of Ga atoms across the close-packed O layers is the rate-determining step and yields “amorphous-like” high energy intermediates, the shearing of O layers contributes to the formation of coherent biphase junctions and the presence of a crystallographic orientation relation, (001)α//(20)β + [120]α//[12]β. Our experiment using high-resolution transmission electron microscopy further confirms the theoretical predictions on the atomic structure of biphase junction and the formation of (20)β twin, and also discovers the late occurrence of lattice expansion in the nascent β phase that grows out from the parent α phase. By distinguishing pseudomartensitic transition from other types of mechanisms, we propose general rules to predict the product crystallinity of solid phase transition. The new knowledge on the kinetics of pseudomartensitic transition complements the theory of diffusionless solid phase transition.
Co-reporter:Shu-Hui Guan and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 6) pp:NaN4534-4534
Publication Date(Web):2016/01/12
DOI:10.1039/C5CP07299A
Structural inhomogeneity is ubiquitous in solid crystals and plays critical roles in phase nucleation and propagation. Here, we develop a heterogeneous solid–solid phase transition theory for predicting the prevailing heterophase junctions, the metastable states governing microstructure evolution in solids. Using this theory and first-principles pathway sampling simulation, we determine two types of heterophase junctions pertaining to metal α–ω phase transition at different pressures and predict the reversibility of transformation only at low pressures, i.e. below 7 GPa. The low-pressure transformation is dominated by displacive Martensitic mechanism, while the high-pressure one is controlled by the reconstructive mechanism. The mechanism of α–ω phase transition is thus highly pressure-sensitive, for which the traditional homogeneous model fails to explain the experimental observations. The results provide the first atomic-level evidence on the coexistence of two different solid phase transition mechanisms in one system.
Co-reporter:Wei-Na Zhao, Sheng-Cai Zhu, Ye-Fei Li and Zhi-Pan Liu
Chemical Science (2010-Present) 2015 - vol. 6(Issue 6) pp:NaN3494-3494
Publication Date(Web):2015/04/07
DOI:10.1039/C5SC00621J
The heterophase solid–solid junction as an important type of structure unit has wide applications for its special mechanics and electronic properties. Here we present a first three-phase atomic model for the anatase–rutile TiO2 heterophase junction and determine its optical and electronic properties, which leads to resolution of the long-standing puzzles on the enhanced photocatalytic activity of anatase–rutile photocatalysts. By using a set of novel theoretical methods, including crystal phase transition pathway sampling, interfacial strain analysis and first principles thermodynamics evaluation of holes and electrons, we identify an unusual structurally ordered three-phase junction, a layer-by-layer “T-shaped” anatase/TiO2-II/rutile junction, for linking anatase with rutile. The intermediate TiO2-II phase, although predicted to be only a few atomic layers thick in contact with anatase, is critical to alleviate the interfacial strain and to modulate photoactivity. We demonstrate that the three-phase junction acts as a single-way valve allowing the photogenerated hole transfer from anatase to rutile but frustrating the photoelectron flow in the opposite direction, which otherwise cannot be achieved by an anatase–rutile direct junction. This new model clarifies the roles of anatase, rutile and the phase junction in achieving high photoactivity synergistically and provides the theoretical basis for the design of better photocatalysts by exploiting multi-phase junctions.
Co-reporter:Wei-Na Zhao and Zhi-Pan Liu
Chemical Science (2010-Present) 2014 - vol. 5(Issue 6) pp:NaN2264-2264
Publication Date(Web):2014/02/13
DOI:10.1039/C3SC53385A
Photocatalytic water splitting is regarded as an important route for generating renewable energy. Here, charged-slab first principles calculations integrated with a periodic continuum solvation model is utilized to analyze the initiating steps of water splitting on the two most common TiO2 surfaces, namely, rutile (110) and anatase (101), at the solid–water interface. It is found that the first proton removal of water (H2O + hole+ → OH + H+) is sensitive to the crystalline phase and surface. The rutile (110) surface is more active for water splitting, with the calculated barrier of O–H bond breaking being 0.2 eV lower compared to that on anatase (101). The higher activity of rutile is not due to the redox level of the hole (the position of the valence band maximum), but caused by the more favorable local bonding geometry of the surface. Unexpectedly, the photogenerated hole does not promote O–H bond breaking, and the charge transfer occurs after the H2O dissociation when the surface O nearby the dissociated OH anion traps the hole. The solvation plays an important catalytic role to stabilize and remove protons from the reaction site, which effectively inhibits the charge-recombination of the dissociated OH anion with the proton. The theory presented here shows that the chemical properties of the surface play a significant role in the photocatalytic process, and a strategy based on simple structural parameters is proposed towards the design of new photocatalysts.
Co-reporter:Qiang Zhao, Lei Li, Fuyou Li, Mengxiao Yu, Zhipan Liu, Tao Yi and Chunhui Huang
Chemical Communications 2008(Issue 6) pp:NaN687-687
Publication Date(Web):2007/11/30
DOI:10.1039/B712416C
A novel aggregation-induced phosphorescent emission (AIPE) was observed for iridium(III) complexes. This interesting phenomenon was attributed to the intermolecular packing, resulting in a switch from the non-emissive 3LX excited state to the emissive 3MLLCT transition, which is confirmed by X-ray diffraction studies as well as theoretical calculations.
Co-reporter:Xiao-Jie Zhang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 4) pp:NaN2769-2769
Publication Date(Web):2014/12/01
DOI:10.1039/C4CP04456H
The prediction of chemical reactivity and thus the design of new reaction systems are the key challenges in chemistry. Here, we develop an unbiased general-purpose reaction sampling method, the stochastic surface walking based reaction sampling (SSW-RS) method, and show that the new method is a promising solution for reactivity prediction of complex reaction systems. The SSW-RS method is capable of sampling both the configuration space of the reactant and the reaction space of pathways, owing to the combination of two recently developed theoretical methods, namely, the stochastic surface walking (SSW) method for potential energy surface (PES) exploration and the double-ended surface walking (DESW) method for building pathways. By integrating with first principles calculations, we show that the SSW-RS method can be applied to investigate the kinetics of complex organic reactions featuring many possible reaction channels and complex hydrogen-bonding networks, as demonstrated here using two examples, epoxypropane hydrolysis in aqueous solution and β-D-glucopyranose decomposition. Our results show that simultaneous sampling of the soft hydrogen-bonding conformations and the chemical reactions involving hard bond making/breaking can be achieved in the SSW-RS simulation, and the mechanism and kinetics can be predicted without a priori information on the system. Unexpected new chemistry for these reactions is revealed and discussed. In particular, despite many possible pathways for β-D-glucopyranose decomposition, the SSW-RS shows that only β-D-glucose and levoglucosan are kinetically preferred direct products and the 5- or 7-member ring products should be secondary products derived from β-D-glucose or levoglucosan. As a general tool for reactivity prediction, the SSW-RS opens a new route for the design of rational reactions.
Co-reporter:Xiao-Jie Zhang, Cheng Shang and Zhi-Pan Liu
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 6) pp:NaN4733-4733
Publication Date(Web):2017/01/09
DOI:10.1039/C6CP06895B
The crystal to amorphous transformation is a common phenomenon in Nature and has important impacts on material properties. Our current knowledge on such complex solid transformation processes is, however, limited because of their slow kinetics and the lack of long-range ordering in amorphous structures. To reveal the kinetics in the amorphization of solids, this work, by developing iterative reaction sampling based on the stochastic surface walking global optimization method, investigates the well-known crystal to amorphous transformation of silica (SiO2) under external pressures, the mechanism of which has long been debated for its non-equilibrium, pressure-sensitive kinetics and complex product components. Here we report for the first time the global potential energy surface (PES) and the lowest energy pathways for α-quartz amorphization from first principles. We show that the pressurization at 15 GPa, the reaction condition, can lift the quartz phase energetically close to the amorphous zone, which thermodynamically initializes the amorphization. More importantly, the large flexibility of Si cation coordination (including four, five and six coordination) results in many kinetically competing routes to more stable dense forms, including the known MI, stishovite, newly-identified MII and TI phases. All these pathways have high barriers due to the local Si–O bond breaking and are mediated by amorphous structures with five-fold Si. This causes simultaneous crystal-to-crystal and crystal-to-amorphous transitions. The high barrier and the reconstructive nature of the phase transition are the key kinetics origin for silica amorphization under pressures.