Co-reporter:Yue Zhou, Na Zhang, Wenjuan Chen, Lijiao Zhao and Rugang Zhong
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 13) pp:9202-9210
Publication Date(Web):01 Mar 2016
DOI:10.1039/C5CP06276D
Protein–protein interactions (PPIs) are fundamental to all biological processes. Recently, the CK2β-derived cyclic peptide Pc has been demonstrated to efficiently antagonize the CK2α/CK2β interaction and strongly affect the phosphorylation of CK2β-dependent CK2 substrate specificity. The binding affinity of Pc to CK2α is destroyed to different extents by two single-point mutations of Tyr188 to Ala (Y188A) and Phe190 to Ala (F190A), which exert negative effects on the inhibitory activity (IC50) of Pc against the CK2α/CK2β interaction from 3.0 μM to 54.0 μM and ≫100 μM, respectively. However, the structural influences of Y188A and F190A mutations on the CK2α–Pc complex remain unclear. In this study, comparative molecular dynamics (MD) simulations, principal component analysis (PCA), domain cross-correlation map (DCCM) analysis and energy calculations were performed on wild type (WT), Y188A mutant, and F190A mutant systems. The results revealed that ordered communications between hydrophobic and polar interactions were essential for CK2α–Pc binding in the WT system. In addition to the loss of the hydrogen bond between Gln36 of CK2α and Gly189 of Pc in the two mutants, the improper recognition mechanisms occurred through different pathways. These pathways included the weakened hydrophobic interactions in the Y188A mutant as well as decreased polar and hydrophobic interactions in the F190A mutant. The energy analysis results qualitatively elucidated the instability of the two mutants and energetic contributions of the key residues. This study not only revealed the structural mechanisms for the decreased binding affinity of Y188A and F190A mutant CK2α–Pc complexes, but also provided valuable clues for the rational design of CK2α/CK2β subunit interaction inhibitors with high affinity and specificity.
Co-reporter:Na Zhang, Hongtao Zhao
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 15) pp:3594-3597
Publication Date(Web):1 August 2016
DOI:10.1016/j.bmcl.2016.06.013
By deconvoluting 238,073 bioactive molecules in the ChEMBL library into extended Murcko ring systems, we identified a set of 2245 ring systems present in at least 10 molecules. These ring systems belong to 2221 clusters by ECFP4 fingerprints with a minimum intracluster similarity of 0.8. Their overlap with ring systems in commercial libraries was further quantified. Our findings suggest that success of a small fragment library is driven by the convergence of effective coverage of bioactive ring systems (e.g., 10% coverage by 1000 fragments vs. 40% by 2 million HTS compounds), high enrichment of bioactive ring systems, and low molecular complexity enhancing the probability of a match with the protein targets. Reconciling with the previous studies, bioactive ring systems are underrepresented in screening libraries. As such, we propose a library of virtual fragments with key functionalities via fragmentation of bioactive molecules. Its utility is exemplified by a prospective application on protein kinase CK2, resulting in the discovery of a series of novel inhibitors with the most potent compound having an IC50 of 0.5 μM and a ligand efficiency of 0.41 kcal/mol per heavy atom.
Co-reporter:Yue Zhou;Xitao Li;Rugang Zhong
Chemical Biology & Drug Design 2015 Volume 85( Issue 2) pp:189-200
Publication Date(Web):
DOI:10.1111/cbdd.12372
Protein kinase CK2 is a novel potential target for cancer treatment. The tricyclic quinoline compound CX-4945 (R2 = COOH) is the first bioavailable CK2 inhibitor used in human clinical trials for advanced solid tumors. CX-4945 analogs with non-R2 carboxylate function were demonstrated to be approximately 5000-fold less potent than compound 12 (R2 = COOH) in vitro. Molecular docking and molecular dynamics simulations were employed to elucidate the structural mechanisms through which the R2 non-ionizable and R3 carboxylic acid substituents influence binding affinity. Results show that the structure of CK2α and the orientation of ligands changed to different degrees in non-R2 carboxylate function systems. The inappropriate electrostatic interactions between the non-R2 carboxylate group and the positive region lead to improper protein–ligand recognition, which is followed by the reorientation of tricyclic skeletons. For CK2α, the affected positions are distributed over the glycine-rich loop (G-loop), C-loop, and the β4/β5 loop. The allosteric mechanisms between the deviated ligands and the changed regions are proposed. Detailed energy calculation and residue-based energy decomposition indicate the energetic influences on the contributions of the critical residues. These results are in accordance with one another and could provide rational clues to the design of more potent CK2 inhibitors.
Co-reporter:Na Zhang;Rugang Zhong;Hong Yan;Yongjun Jiang
Chemical Biology & Drug Design 2011 Volume 77( Issue 3) pp:199-205
Publication Date(Web):
DOI:10.1111/j.1747-0285.2010.01069.x
Hymenialdisine and dibromocantharelline are marine sponge constituents with unique biological activity. Hymenialdisine potently inhibits glycogen synthase kinase 3β, cyclin-dependent kinase 2, and cyclin-dependent kinase 5, whereas dibromocantharelline only displays a significant inhibitory effect toward glycogen synthase kinase 3β (IC50 = 3 μmol). Based on the crystal structure of cyclin-dependent kinase 2–hymenialdisine complex, we employed three docking methods, namely Autodock, FlexX, and Genetic Optimization for Ligand Docking, as well as molecular dynamics simulations to investigate the structural determinants that govern target selectivity. The similar binding modes of hymenialdisine in complex with cyclin-dependent kinase 5 and glycogen synthase kinase 3β are consistent with the poor selectivity of hymenialdisine toward the two kinases. The shape of cyclin-dependent kinase 5 binding pocket characterized by the inward-orientation of Asp144 and dense electrostatic interaction networks, as well as the stereochemical configuration of dibromocantharelline, provides a considerable structural basis for the lack of binding of dibromocantharelline with cyclin-dependent kinase 5. The specific residue Cys199 near the binding site of glycogen synthase kinase 3β provides new clues for the design of potent and selective inhibitor of glycogen synthase kinase 3β.
Co-reporter:Na Zhang, Rugang Zhong
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 1) pp:292-297
Publication Date(Web):January 2010
DOI:10.1016/j.ejmech.2009.10.011
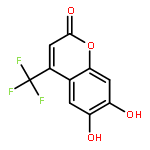
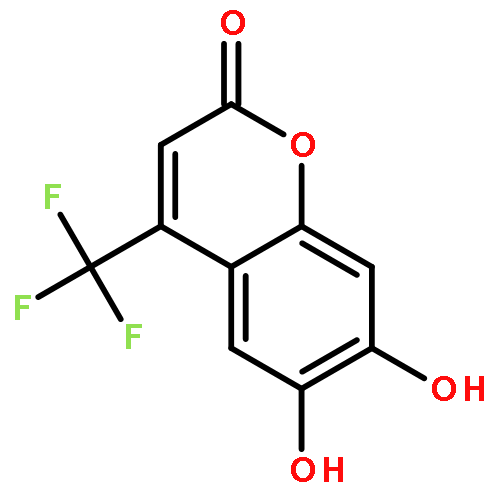
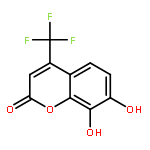
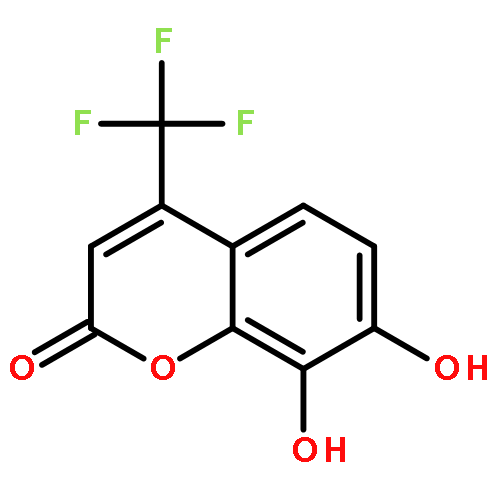
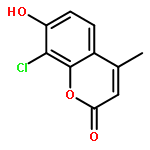
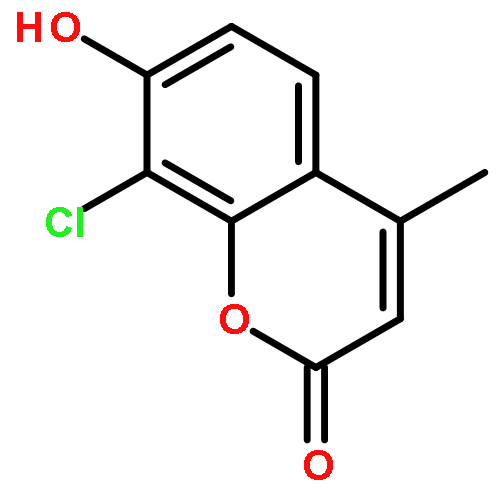
Protein kinase CK2 is involved in a broad range of physiological events. 3,8-dibromo-7-hydroxy-4-methylchromen-2-one (DBC) analogues show favorable inhibitory activity targeting CK2α. Two methods were used to build 3D-QSAR models for DBC derivatives. The ligand-based (LB) studies were performed based on the lower energy conformations employing atom fit alignment rule. The receptor-based (RB) models were also derived using bioactive conformations. Contour maps of RB CoMSIA model (q2 = 0.694, r2 = 0.916, N (no. of components) = 7, r2pred = 0.87) including the steric, electronic, hydrophobic and hydrogen bond acceptor fields were employed to explain factors affecting activities of inhibitors. The good consistency between the contour maps and the properties of CK2α amino acids provide useful hints for new inhibitors design.The 3D QSAR study of CK2 inhibition by 7-hydroxycoumarin derivatives based on two methods (ligand-based and receptor-based) is reported.
Co-reporter:Na Zhang;Rugang Zhong
Journal of Molecular Modeling 2010 Volume 16( Issue 4) pp:771-780
Publication Date(Web):2010 April
DOI:10.1007/s00894-009-0582-2
Protein kinase CK2 (casein kinase 2) is a multifunctional serine/threonine kinase that is involved in a broad range of physiological events. The decreased affinity of Emodin binding to human CK2α resulting from single-point mutation of Val66 to Ala (V66A) has been demonstrated by experimental mutagenesis. Molecular dynamics (MD) simulations and energy analysis were performed on wild type (WT) and V66A mutant CK2α-Emodin complexes to investigate the subtle influences of amino acid replacement on the structure of the complex. The structure of CK2α and the orientation of Emodin undergo changes to different degrees in V66A mutant. The affected positions in CK2α are mainly distributed over the glycine-rich loop (G-loop), the α-helix and the loop located at the portion between G-loop and α-helix (C-loop). Based on the coupling among these segments, an allosteric mechanism among the C-loop, the G-loop and the deviated Emodin is proposed. Additionally, an estimated energy calculation and residue-based energy decomposition also indicate the lower instability of V66A mutant in contrast to WT, as well as the unfavorable energetic influences on critical residue contributions.
Co-reporter:Yue Zhou, Na Zhang, Wenjuan Chen, Lijiao Zhao and Rugang Zhong
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 13) pp:NaN9210-9210
Publication Date(Web):2016/03/01
DOI:10.1039/C5CP06276D
Protein–protein interactions (PPIs) are fundamental to all biological processes. Recently, the CK2β-derived cyclic peptide Pc has been demonstrated to efficiently antagonize the CK2α/CK2β interaction and strongly affect the phosphorylation of CK2β-dependent CK2 substrate specificity. The binding affinity of Pc to CK2α is destroyed to different extents by two single-point mutations of Tyr188 to Ala (Y188A) and Phe190 to Ala (F190A), which exert negative effects on the inhibitory activity (IC50) of Pc against the CK2α/CK2β interaction from 3.0 μM to 54.0 μM and ≫100 μM, respectively. However, the structural influences of Y188A and F190A mutations on the CK2α–Pc complex remain unclear. In this study, comparative molecular dynamics (MD) simulations, principal component analysis (PCA), domain cross-correlation map (DCCM) analysis and energy calculations were performed on wild type (WT), Y188A mutant, and F190A mutant systems. The results revealed that ordered communications between hydrophobic and polar interactions were essential for CK2α–Pc binding in the WT system. In addition to the loss of the hydrogen bond between Gln36 of CK2α and Gly189 of Pc in the two mutants, the improper recognition mechanisms occurred through different pathways. These pathways included the weakened hydrophobic interactions in the Y188A mutant as well as decreased polar and hydrophobic interactions in the F190A mutant. The energy analysis results qualitatively elucidated the instability of the two mutants and energetic contributions of the key residues. This study not only revealed the structural mechanisms for the decreased binding affinity of Y188A and F190A mutant CK2α–Pc complexes, but also provided valuable clues for the rational design of CK2α/CK2β subunit interaction inhibitors with high affinity and specificity.



![1H-Indazole-3-carboxamide, N-[3-(methylsulfonyl)phenyl]-](http://img.cochemist.com/ccimg/660900/660822-46-8.png)
![1H-Indazole-3-carboxamide, N-[3-(methylsulfonyl)phenyl]-](http://img.cochemist.com/ccimg/660900/660822-46-8_b.png)
![8-butyl-1-methyl-7-phenyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione](http://img.cochemist.com/ccimg/85600/85592-05-8.png)
![8-butyl-1-methyl-7-phenyl-1H-imidazo[2,1-f]purine-2,4(3H,8H)-dione](http://img.cochemist.com/ccimg/85600/85592-05-8_b.png)