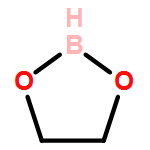
Co-reporter:Chao Wu, Xiufang Hou, Yuheng Zheng, Pengfei Li, and Dongmei Lu
The Journal of Organic Chemistry March 17, 2017 Volume 82(Issue 6) pp:2898-2898
Publication Date(Web):February 22, 2017
DOI:10.1021/acs.joc.6b02849
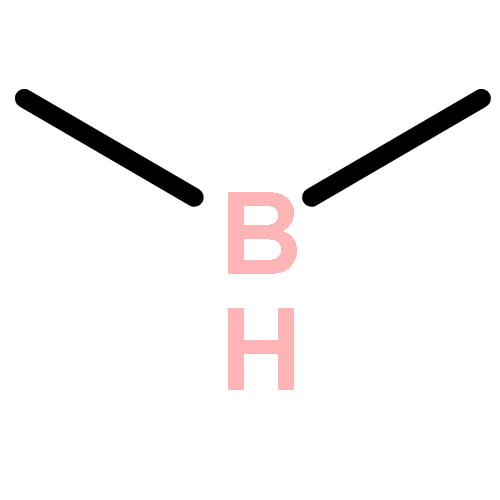
We carried out a survey of the relative reactivity of a collection of 91 neutral boryl radicals using density functional calculations. Their reactivities were characterized by four indices, i.e., the global electrophilicity, global nucleophilicity, local electrophilicity, and local nucleophilicity. Particularly, the global electrophilicity and nucleophilicity indices span over a moderately wider range than those of carbon radicals, indicating their potentially broader reactivity. Thus, boryl radicals may be utilized in electrophilic radical reactions, while traditionally they are only considered for nucleophilic radical reactions. In contrast, the local electrophilicity and nucleophilicity indices at the boron center show a different reactivity picture than their global counterparts. The inconsistency is rooted in the low and varying spin density on boron (for most radicals, less than 50%) in different boryl radicals, which is a combinative result of radical stabilization via electron delocalization and the low electronegativity of boron (compared to carbon). In short, the boron character in boryl radicals may be weak and their reactivity is not reflected by the local indices based on boron but by the global ones.
Co-reporter:Xiufang Hou, Dongmei Lu, Ru Liu
Computational and Theoretical Chemistry 2016 Volume 1092() pp:25-31
Publication Date(Web):15 September 2016
DOI:10.1016/j.comptc.2016.07.024
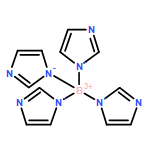
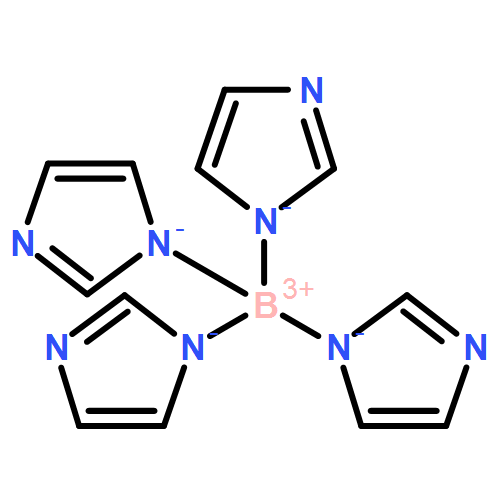
•A group of poly(azolyl)borane radical anions ([B(az)(n)H(3−n)]−, az = azolyl group, n = 1, 2, 3).•Poly(azolyl)borane radical anions electronic structure (spin distribution) and reactivity with H atoms and azolyl radicals.•The stabilizing capability of the unpaired electron by the poly(azolyl)borane scaffolds.•Be comparable to the synthesized borane radical derived from borole and triphenylborane ([BR3]−).•They can be used as precursors to develop a unique family of boron-centered radical anions.A group of poly(azolyl)borane radical anions (B(az)nH3−n−; az = azolyl group; n = 1, 2, 3) have been proposed and theoretically studied. When one of the BH bond(s) of a hydropoly(azolyl)borate anion homolytically dissociates, the corresponding radical anion is formed, whose BH bond dissociation energy (BDE) lies moderately higher (by ∼30 to 40 kJ/mol) than that of Lewis base-stabilized (LB = amines, nitrogen heterocycles, carbenes, etc.) boranes, which have been experimentally converted into neutral boryl radicals (BR2LB). Unlike boryl radicals, the BH BDEs of the proposed borane radical anions do not have an evident linear correlation with the spin population on boron, which is a result of the complex electronic and steric effects caused by the versatile and complex structures of azolyl groups and their degree of substitutions (i.e., mono-, bi-, or tri-). Essentially, the stabilizing capability of the unpaired electron by poly(azolyl)borane scaffolds are comparable to the synthesized borane radical anions derived from borole and triphenylborane (BR3−), suggesting that the well-studied ligand family of poly(azolyl)borates may be precursors to develop a unique family of boron-centered radical anions.A group of borane radical anions utilizing the poly(azolyl)borate scaffold are proposed.
Co-reporter:Dongmei Lu and Huarong Tang
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 26) pp:17027-17033
Publication Date(Web):02 Jun 2015
DOI:10.1039/C5CP02094H
Using density functional calculations, we have systematically investigated a series of homoleptic poly(azolyl)borate ligands, which display unusual steadily declining bond strengths accompanied by bond contractions when the azolyl groups are sequentially substituted to the parent BH4−. As ligands, their effects on the coordinated metal ions (Cu(I) and Mo(0)) are quantitatively represented by two ligand tunability descriptors: the vibration frequency (νCO) of the CO groups complexed to the metal ions and the charge of the metal–(CO)x moiety, between which a good linear correlation exists. For the same number of azolyl substitutions, the boundary of ligand tunability is always marked by the pyrazolyl and tetrazolyl groups at the two ends, which feature the lowest and the highest nitrogen content in the azolyl ring, respectively. With the increase of the azolyl substitution number in the borate ligands, the νCO range expands, indicating a higher tunability of the ligands. The type of metal ion and the charge they carry play minor roles in influencing the ligand tunability.
Co-reporter:Huarong Tang, Dongmei Lu and Chao Wu
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 24) pp:15725-15731
Publication Date(Web):12 May 2015
DOI:10.1039/C5CP01793A
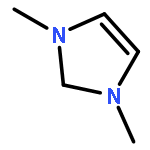
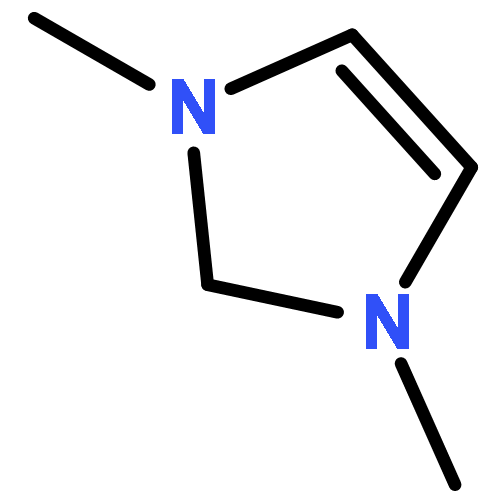
Cation-assisted interactions between N-containing heterocycles (NHCs) and CO2 have been systematically studied by using density functional theory (DFT). For neutral and anionic (non-carbenoid) NHCs, the effects of monovalent cations (i.e., alkali metal ions) are moderate to small (the NHC–CO2 binding energy change, ΔBE usually < 25 kJ mol−1). However, for NHC carbenes, due to their strong basicity, the effects are strong (ΔBE > 60 kJ mol−1) and the monovalent cations play a critical role in the single carboxylation of dicarbenes with CO2. In comparison, divalent alkali earth metal cations, due to both their smaller sizes and higher formal charges, exhibit a much stronger influence (ΔBE > 100 kJ mol−1). Divalent cations should be incorporated into next generation CO2 capture reagents. Other aspects including the reaction potential energy surface (PES), orbital-based analyses of interactions, substitution effects, and the reactivity descriptors (cation size, reacting N lone pair orbital energy, etc.) have been discussed in detail as well.
Co-reporter:Huarong Tang;Dr. Dongmei Lu;Dr. Chao Wu
ChemPhysChem 2015 Volume 16( Issue 9) pp:1926-1932
Publication Date(Web):
DOI:10.1002/cphc.201500164
Abstract
Reducing the emission of greenhouse gases, such as CO2, requires efficient and reusable capture materials. The energy for regenerating sorbents is critical to the cost of CO2 capture. Here, we design a series of photoswitchable CO2 capture molecules by grafting Lewis bases, which can covalently bond CO2, to azo-based backbones that can switch configurations upon light stimulation. The first-principles calculations demonstrate that intramolecular hydrogen bonds are crucial for enlarging the difference of CO2 binding strengths to the cis and trans isomers. As a result, the CO2–sorbent interaction can be light-adjusted from strong chemical bonding in one configuration to weak bonding in the other, which may lead to a great energy reduction in sorbent regeneration.
Co-reporter:Huarong Tang;Dr. Dongmei Lu
ChemPhysChem 2015 Volume 16( Issue 13) pp:2854-2860
Publication Date(Web):
DOI:10.1002/cphc.201500369
Abstract
We propose a series of azolium poly(azolyl)borate ionic liquids (ILs) for reversible SO2 capture. Density functional calculations demonstrate that the designed borate anions can strongly bond to SO2 at multiple sites with nearly uniform binding energies. Thus, as well as high overall uptakes, the ILs can achieve much higher effective uptakes (the uptake difference between absorption and desorption conditions) than existing SO2-capture reagents. The larger size of the borate anions, the evenly distributed negative charge among the azolyl rings, and the blocking of the conjugation by the tetrahedral boron concertedly reduce absorbate–absorbate repulsion, which leads to a large disparity among binding sites in other multiple-site SO2 sorbents.
Co-reporter:Dongmei Lu, Chao Wu, and Pengfei Li
Organic Letters 2014 Volume 16(Issue 5) pp:1486-1489
Publication Date(Web):February 19, 2014
DOI:10.1021/ol500296a
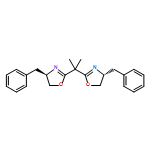
Five- and six-membered boron heterocycle-based three-center-five-electron (3c–5e) type boryl radicals with unusual σ0π1 ground state electronic structures are predicted theoretically. Compared to σ1π0 analogs, their unique electronic structure leads to both lower reactivity toward H-atoms and stronger coordination with Lewis bases. The corresponding Lewis base-stabilized four-center-seven-electron (4c–7e) type boryl radicals are even more unreactive toward H-atoms than the conventional 4c–7e ones.
Co-reporter:Dr. Dongmei Lu;Dr. Chao Wu;Dr. Pengfei Li
Chemistry - A European Journal 2014 Volume 20( Issue 6) pp:1630-1637
Publication Date(Web):
DOI:10.1002/chem.201303705
Abstract
Boryl radicals have the potential for the development of new molecular entities and for application in new radical reactions. However, the effects of the substituents and coordinating Lewis bases on the reactivity of boryl radicals are not fully understood. By using first-principles methods, we investigated the spin-density distribution and reactivity of a series of boryl radicals with various substituents and Lewis bases. The substituents, along with the Lewis bases, only affect the radical reactivity when an unpaired electron is in the boron pz orbital, that is, for three-coordinate radicals. We found evidence of synergistic effects between the substituents and the Lewis bases that can substantially broaden the tunability of the reactivity of the boryl radicals. Among Lewis bases, pyridine and imidazol-2-ylidene show a similar capacity for stabilization by delocalizing the spin density. Electron-donating substituents, such as nitrogen, more efficiently stabilize boryl radicals than oxygen and carbon atoms. The reactivity of a boryl radical is always boron based, irrespective of the spin density on boron.
Co-reporter:Dongmei Lu and Huarong Tang
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 26) pp:NaN17033-17033
Publication Date(Web):2015/06/02
DOI:10.1039/C5CP02094H
Using density functional calculations, we have systematically investigated a series of homoleptic poly(azolyl)borate ligands, which display unusual steadily declining bond strengths accompanied by bond contractions when the azolyl groups are sequentially substituted to the parent BH4−. As ligands, their effects on the coordinated metal ions (Cu(I) and Mo(0)) are quantitatively represented by two ligand tunability descriptors: the vibration frequency (νCO) of the CO groups complexed to the metal ions and the charge of the metal–(CO)x moiety, between which a good linear correlation exists. For the same number of azolyl substitutions, the boundary of ligand tunability is always marked by the pyrazolyl and tetrazolyl groups at the two ends, which feature the lowest and the highest nitrogen content in the azolyl ring, respectively. With the increase of the azolyl substitution number in the borate ligands, the νCO range expands, indicating a higher tunability of the ligands. The type of metal ion and the charge they carry play minor roles in influencing the ligand tunability.
Co-reporter:Huarong Tang, Dongmei Lu and Chao Wu
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 24) pp:NaN15731-15731
Publication Date(Web):2015/05/12
DOI:10.1039/C5CP01793A
Cation-assisted interactions between N-containing heterocycles (NHCs) and CO2 have been systematically studied by using density functional theory (DFT). For neutral and anionic (non-carbenoid) NHCs, the effects of monovalent cations (i.e., alkali metal ions) are moderate to small (the NHC–CO2 binding energy change, ΔBE usually < 25 kJ mol−1). However, for NHC carbenes, due to their strong basicity, the effects are strong (ΔBE > 60 kJ mol−1) and the monovalent cations play a critical role in the single carboxylation of dicarbenes with CO2. In comparison, divalent alkali earth metal cations, due to both their smaller sizes and higher formal charges, exhibit a much stronger influence (ΔBE > 100 kJ mol−1). Divalent cations should be incorporated into next generation CO2 capture reagents. Other aspects including the reaction potential energy surface (PES), orbital-based analyses of interactions, substitution effects, and the reactivity descriptors (cation size, reacting N lone pair orbital energy, etc.) have been discussed in detail as well.







![3H-Imidazo[4,5-b]pyridin-7-amine](http://img.cochemist.com/ccimg/6800/6703-44-2.png)
![3H-Imidazo[4,5-b]pyridin-7-amine](http://img.cochemist.com/ccimg/6800/6703-44-2_b.png)