Co-reporter:S. J. Coles;L. K. Mapp
Journal of Chemical Education 2016 Volume 93(Issue 1) pp:131-140
Publication Date(Web):November 5, 2015
DOI:10.1021/acs.jchemed.5b00071
An undergraduate practical exercise has been designed to provide hands-on, instrument-based experience of advanced characterization techniques. A research experience approach is taken, centered around the concept of solid-state polymorphism, which requires a detailed knowledge of molecular and crystal structure to be gained by advanced analytical techniques normally considered as the preserve of a research facility. Powder and single crystal diffraction techniques are primarily required and implemented via the unique approach of the students themselves using benchtop instruments dedicated to teaching, as opposed to more complex and difficult to access research instruments. Furthermore, the manual instructions for performing the practical are delivered via an adapted Electronic Laboratory Notebook system where, for each specific aspect of the practical, students note their intentions, actions, observations, and inferences. Assessors can access the notebooks and provide targeted online feedback for each individual section. Evaluation of the approach is based on interviews and surveys with the first cohort of 65 students that performed the practical.
Co-reporter:Rebecca K. Hylton; Graham J. Tizzard; Terence L. Threlfall; Amy L. Ellis; Simon J. Coles; Colin C. Seaton+; Eric Schulze; Heike Lorenz; Andreas Seidel-Morgenstern; Matthias Stein;Sarah L. Price
Journal of the American Chemical Society 2015 Volume 137(Issue 34) pp:11095-11104
Publication Date(Web):August 5, 2015
DOI:10.1021/jacs.5b05938
Mandelic acids are prototypic chiral molecules where the sensitivity of crystallized forms (enantiopure/racemic compound/polymorphs) to both conditions and substituents provides a new insight into the factors that may allow chiral separation by crystallization. The determination of a significant number of single crystal structures allows the analysis of 13 enantiopure and 30 racemic crystal structures of 21 (F/Cl/Br/CH3/CH3O) substituted mandelic acid derivatives. There are some common phenyl packing motifs between some groups of racemic and enantiopure structures, although they show very different hydrogen-bonding motifs. The computed crystal energy landscape of 3-chloromandelic acid, which has at least two enantiopure and three racemic crystal polymorphs, reveals that there are many more possible structures, some of which are predicted to be thermodynamically more favorable as well as slightly denser than the known forms. Simulations of mandelic acid dimers in isolation, water, and toluene do not differentiate between racemic and enantiopure dimers and also suggest that the phenyl ring interactions play a major role in the crystallization mechanism. The observed crystallization behavior of mandelic acids does not correspond to any simple “crystal engineering rules” as there is a range of thermodynamically feasible structures with no distinction between the enantiopure and racemic forms. Nucleation and crystallization appear to be determined by the kinetics of crystal growth with a statistical bias, but the diversity of the mandelic acid crystallization behavior demonstrates that the factors that influence the kinetics of crystal nucleation and growth are not yet adequately understood.
Co-reporter:L. H. Blair, A. Colakel, R. M. Vrcelj, I. Sinclair and S. J. Coles
Chemical Communications 2015 vol. 51(Issue 61) pp:12185-12188
Publication Date(Web):23 Jun 2015
DOI:10.1039/C5CC04174K
A new approach to formulating pyrotechnic materials is presented whereby constituent ingredients are bound together in a solid-state lattice. This reduces the batch inconsistencies arising from the traditional approach of combining powders by ensuring the key ingredients are ‘mixed’ in appropriate quantities and are in intimate contact. Further benefits of these types of material are increased safety levels as well as simpler logistics, storage and manufacture. A systematic series of new frameworks comprising fuel and oxidiser agents (group 1 and 2 metal nodes & terephthalic acid derivatives as linkers) has been synthesised and structurally characterised. These new materials have been assessed for pyrotechnic effect by calorimetry and burn tests. Results indicate that these materials exhibit the desired pyrotechnic material properties and the effect can be correlated to the dimensionality of the structure. A new approach to formulating pyrotechnic materials is proposed whereby constituent ingredients are bound together in a solid-state lattice. A series of Metal–organic framework frameworks comprising fuel and oxidiser agents exhibits the desired properties of a pyrotechnic material and this effect is correlated to the dimensionality of the structure.
Co-reporter:Isabelle L. Kirby, Mateusz B. Pitak, Claire Wilson, Philip A. Gale and Simon J. Coles
CrystEngComm 2015 vol. 17(Issue 14) pp:2815-2826
Publication Date(Web):25 Feb 2015
DOI:10.1039/C5CE00213C
Building on previous studies of anion-receptor complexes based on a urea scaffold substituted symmetrically with electron-withdrawing nitro groups, the electron density distribution in an analogous thiourea receptor complex and the related asymmetrically substituted urea and thiourea receptors are described. On this basis it is possible to probe both the effect of changing the receptor core from a urea to a thiourea moiety and that of asymmetrical substitution of the receptor molecule. These modifications are shown to significantly alter the anion binding properties, solid-state packing and electron density distribution in the anion-receptor complexes.
Co-reporter:Isabelle L. Kirby, Mark Brightwell, Mateusz B. Pitak, Claire Wilson, Simon J. Coles and Philip A. Gale
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 22) pp:10943-10958
Publication Date(Web):28 Apr 2014
DOI:10.1039/C3CP54858A
The first systematic electronic resolution study of a series of urea-based anion receptor complexes is presented. The hydrogen bonding in these multi-component systems was fully characterised using Bader's Quantum Theory of Atoms In Molecules (QTAIM) with the strength of the various N–H⋯anion hydrogen bonds quantified and the individual contributions of different intermolecular forces to the overall receptor: anion interaction derived by comparison of the charge densities in the related complexes. The strength of the N–H⋯anion hydrogen bonds was correlated to the basicity of the anion and related to the structure of the receptors. The geometric criteria used to identify hydrogen bonding interactions in standard resolution X-ray diffraction studies were shown to be valid for stronger interactions. However, these geometric criteria are less reliable and lead to assumptions that are not necessarily upheld when applied to weaker intermolecular interactions. The presence of these could only be confirmed by charge density studies. The effect that changes to the receptor substitution pattern have on the entire supramolecular system is illustrated by the differences in the electrostatic potential distributions and atomic charges across the series. The application of systematic high resolution studies to rationalise a variety of host–guest systems has been demonstrated.
Co-reporter:Simon J. Coles, Terence L. Threlfall, and Graham J. Tizzard
Crystal Growth & Design 2014 Volume 14(Issue 4) pp:1623-1628
Publication Date(Web):February 20, 2014
DOI:10.1021/cg401655h
The expression “isostructural polymorphs” would appear to be an impossible combination of two mutually incompatible words. “Isostructural” implies a high degree of structural similarity; however, conversely, “polymorph” implies structural distinguishability. The structures of two newly determined polymorphs of 3-chloromandelic acid nevertheless justify the use of this expression, for they differ only in crystal symmetry and hardly at all in molecular position or conformation within the crystalline lattice. We demonstrate that parameters derived by the XPac program can be useful in establishing the limits of isostructurality.
Co-reporter:S. J. Coles, A. L. Ellis, K. Leung, J. Sarson, T. L. Threlfall and G. J. Tizzard
CrystEngComm 2014 vol. 16(Issue 47) pp:10816-10823
Publication Date(Web):14 Oct 2014
DOI:10.1039/C4CE01832J
The structures of 27 monosubstituted mandelic acids, including several of their polymorphs, plus unsubstituted mandelic acid itself (two polymorphs) are investigated for structural similarity. The results, presented pictorially as a structural relationship plot, show that rather more structures are built up from the carboxyl-chain hydroxyl hydrogen bonded dimer than from the conventional carboxylic acid dimer. The results show how all the structures are related and, based on the two types of dimer, the degree of similarity that they possess. Some structures with Z′ > 1 contain both sorts of dimers and there are many examples of isostructural sets within the structures so far determined. We also present an example where analysing similarity in related families of structures highlights a structure that should be present and which has indeed then proceeded to be synthesised and determined.
Co-reporter:Isabelle L. Kirby, Mateusz B. Pitak, Marco Wenzel, Claire Wilson, Hazel A. Sparkes, Simon J. Coles and Philip A. Gale
CrystEngComm 2013 vol. 15(Issue 44) pp:9003-9010
Publication Date(Web):29 Aug 2013
DOI:10.1039/C3CE41503A
A crystallographic systematic study of a series of 6 anion receptor complexes has been performed, with the accurate atomic positions and displacement parameters of the hydrogen atoms for two complexes determined by neutron diffraction studies. The N–H⋯anion hydrogen bonding interaction is shown to be central to the geometry of the urea unit. Contributions of the intermolecular interactions to the packing of the molecule are highlighted by correlation to the Hirshfeld surfaces and their fingerprint plots, with alterations to the position and amount of nitro substitution on the receptor shown to affect the π⋯π stacking motifs observed across the structures.
Co-reporter:Simon J. Coles and Philip A. Gale
Chemical Science 2012 vol. 3(Issue 3) pp:683-689
Publication Date(Web):09 Dec 2011
DOI:10.1039/C2SC00955B
Crystallography is no longer solely the preserve of the specialist, a situation that has implications for the operation of crystallographic service facilities. This mini-review provides an overview of the challenges in operating a crystallographic facility from the perspective and experience of the UK National Crystallography Service – a modern mid-range facility. Examples of chemical research generating the greatest challenges for the modern crystallography service and the state-of-the-art tools, hardware, facilities and expertise that are required to address them are highlighted. An overview of current research trends in single crystal diffraction research, which will ensure the future development of the technique, is presented. The remit of the service crystallographer is examined, together with proposed examples of best practice.
Co-reporter:Ferda Hacıvelioğlu, Riccardo Montis, David B. Davies, Adem Kılıç, Michael B. Hursthouse and Simon J. Coles
CrystEngComm 2011 vol. 13(Issue 12) pp:4102-4109
Publication Date(Web):15 Apr 2011
DOI:10.1039/C1CE05163F
Conformational polymorphism has previously been observed in 2,2-dichloro-4,4:6,6-bis(2,2′-dimethlypropane-1,3-dioxy)-cyclotriphosphazene and the crystal structures of two forms were reported. In that study the melting points of both forms (α and β) were reported as 211–212 °C and no investigation was made on the stability of, or the thermodynamic relationships between the polymorphs. In the present work the two forms were characterized by various analytical techniques, such as single-crystal X-ray diffraction, differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), thermomicroscopy (TM) and variable temperature single crystal and powder X-ray diffraction. Thermal analysis suggests that the polymorphs are enantiotropically related and it is found that the solid–solid transformation of the β to α form may start with defects of α form inside the β form followed by a nucleation and growth type mechanism involving at least three main molecular centred events; conformational change, rotation and tilting.
Co-reporter:S. J. Coles, T. Gelbrich, U. J. Griesser, M. B. Hursthouse, M. Pitak and T. Threlfall
Crystal Growth & Design 2009 Volume 9(Issue 11) pp:4610-4612
Publication Date(Web):October 15, 2009
DOI:10.1021/cg901045t
Co-reporter:David W. Allen, Simon J. Coles, Mark E. Light, Michael B. Hursthouse
Inorganica Chimica Acta 2004 Volume 357(Issue 5) pp:1558-1564
Publication Date(Web):25 March 2004
DOI:10.1016/j.ica.2003.12.001
The synthesis of tri(2-furyl)(8-quinolylmethyl)phosphonium bromide and 2-[2-tri(2-furyl)phosphoniophenyl]benzimidazole perchlorate is described, the latter involving a nickel(II)-catalysed displacement of bromine from 2-(2-bromophenyl)benzimidazole by tri(2-furyl)phosphine. X-ray structural studies of the phosphoniobenzimidazole salt reveals the existence of a significant hypervalent coordinative interaction between heterocyclic nitrogen and the phosphonium centre, which also appears to be retained in solution, the 31P NMR spectrum showing a significantly shielded phosphorus atom, δ31P=ca. 40 ppm in CDCl3. The structure of the phosphoniophenylbenzimidazole cation reveals major distortion of bond angles about phosphorus away from the idealised tetrahedral angles expected for a tetraarylphosphonium salt, in the range 102–116°. Three of the angles are reduced below the tetrahedral angle and three are increased, the structure about phosphorus approaching that of a trigonal bipyramid, in which the heterocyclic imino nitrogen forms part of a five-membered ring spanning apical–equatorial positions. The apical axis of the trigonal bipyramid is formed by this nitrogen atom and one of the 2-furyl groups, the apical axial bond angle (N2–P–C14) being an average of 178°. The remaining 2-furyl groups occupy equatorial positions, along with the phenyl ring. Significantly, the nitrogen–phosphorus distance is an average of 2.67 Å (for two independent molecules in the unit cell), being the shortest observed in structures of this type, a consequence of the electron-withdrawing properties of the 2-furyl substituents at phosphorus. The structure also shows edge to face associations of 2-furyl substituents of one cation with the phenyl ring of the benzimidazole unit of another cation. The perchlorate anion is hydrogen-bonded to the nitrogen bearing the hydrogen atom in the benzimidazole ring system. In contrast, the N–P interaction in the quinolylmethylphosphonium salt is much less developed, with an N–P distance of 3.511 Å, although there is considerable deformation of bond angles at phosphorus. The crystal structure is dominated by the existence of hydrogen-bonded interactions between the cation, anion and a molecule of water, and by face to face interactions between cations. Both salts undergo loss of a 2-furyl group on treatment with hydroxide ion.The crystal structure of the tri(2-furyl)phosphonio-benzimidazole cation reveals a significant hypervalent coordinative interaction between a heterocyclic nitrogen atom and the phosphonium centre, which appears to be retained in solution, the 31P NMR spectrum showing a significantly shielded phosphorus atom, δ31P=ca.−40 ppm in CDCl3.
Co-reporter:Simon J. Coles and Philip A. Gale
Chemical Science (2010-Present) 2012 - vol. 3(Issue 3) pp:NaN689-689
Publication Date(Web):2011/12/09
DOI:10.1039/C2SC00955B
Crystallography is no longer solely the preserve of the specialist, a situation that has implications for the operation of crystallographic service facilities. This mini-review provides an overview of the challenges in operating a crystallographic facility from the perspective and experience of the UK National Crystallography Service – a modern mid-range facility. Examples of chemical research generating the greatest challenges for the modern crystallography service and the state-of-the-art tools, hardware, facilities and expertise that are required to address them are highlighted. An overview of current research trends in single crystal diffraction research, which will ensure the future development of the technique, is presented. The remit of the service crystallographer is examined, together with proposed examples of best practice.
Co-reporter:L. H. Blair, A. Colakel, R. M. Vrcelj, I. Sinclair and S. J. Coles
Chemical Communications 2015 - vol. 51(Issue 61) pp:NaN12188-12188
Publication Date(Web):2015/06/23
DOI:10.1039/C5CC04174K
A new approach to formulating pyrotechnic materials is presented whereby constituent ingredients are bound together in a solid-state lattice. This reduces the batch inconsistencies arising from the traditional approach of combining powders by ensuring the key ingredients are ‘mixed’ in appropriate quantities and are in intimate contact. Further benefits of these types of material are increased safety levels as well as simpler logistics, storage and manufacture. A systematic series of new frameworks comprising fuel and oxidiser agents (group 1 and 2 metal nodes & terephthalic acid derivatives as linkers) has been synthesised and structurally characterised. These new materials have been assessed for pyrotechnic effect by calorimetry and burn tests. Results indicate that these materials exhibit the desired pyrotechnic material properties and the effect can be correlated to the dimensionality of the structure. A new approach to formulating pyrotechnic materials is proposed whereby constituent ingredients are bound together in a solid-state lattice. A series of Metal–organic framework frameworks comprising fuel and oxidiser agents exhibits the desired properties of a pyrotechnic material and this effect is correlated to the dimensionality of the structure.
Co-reporter:Isabelle L. Kirby, Mark Brightwell, Mateusz B. Pitak, Claire Wilson, Simon J. Coles and Philip A. Gale
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 22) pp:NaN10958-10958
Publication Date(Web):2014/04/28
DOI:10.1039/C3CP54858A
The first systematic electronic resolution study of a series of urea-based anion receptor complexes is presented. The hydrogen bonding in these multi-component systems was fully characterised using Bader's Quantum Theory of Atoms In Molecules (QTAIM) with the strength of the various N–H⋯anion hydrogen bonds quantified and the individual contributions of different intermolecular forces to the overall receptor: anion interaction derived by comparison of the charge densities in the related complexes. The strength of the N–H⋯anion hydrogen bonds was correlated to the basicity of the anion and related to the structure of the receptors. The geometric criteria used to identify hydrogen bonding interactions in standard resolution X-ray diffraction studies were shown to be valid for stronger interactions. However, these geometric criteria are less reliable and lead to assumptions that are not necessarily upheld when applied to weaker intermolecular interactions. The presence of these could only be confirmed by charge density studies. The effect that changes to the receptor substitution pattern have on the entire supramolecular system is illustrated by the differences in the electrostatic potential distributions and atomic charges across the series. The application of systematic high resolution studies to rationalise a variety of host–guest systems has been demonstrated.












![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, 4-chloropentyl ester](http://img.cochemist.com/ccimg/853000/852919-53-0.png)
![Bicyclo[2.2.1]hept-5-ene-2-carboxylic acid, 4-chloropentyl ester](http://img.cochemist.com/ccimg/853000/852919-53-0_b.png)


![Silane, trimethyl[2-methylene-1-(phenylmethyl)cyclopropyl]-](http://img.cochemist.com/ccimg/97800/97778-18-2.png)
![Silane, trimethyl[2-methylene-1-(phenylmethyl)cyclopropyl]-](http://img.cochemist.com/ccimg/97800/97778-18-2_b.png)
![2H-Indol-2-one, 3-[(4-bromophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/90900/90828-11-8.png)
![2H-Indol-2-one, 3-[(4-bromophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/90900/90828-11-8_b.png)
![2-Pentanone, 5-[2-methylene-1-(trimethylsilyl)cyclopropyl]-](http://img.cochemist.com/ccimg/263600/263565-94-2.png)
![2-Pentanone, 5-[2-methylene-1-(trimethylsilyl)cyclopropyl]-](http://img.cochemist.com/ccimg/263600/263565-94-2_b.png)
![Bicyclo[3.1.1]heptan-2-ol, 2,6,6-trimethyl-3-phenoxy-, (1R,2R,3S,5R)-](http://img.cochemist.com/ccimg/505100/505030-30-8.png)
![Bicyclo[3.1.1]heptan-2-ol, 2,6,6-trimethyl-3-phenoxy-, (1R,2R,3S,5R)-](http://img.cochemist.com/ccimg/505100/505030-30-8_b.png)






![2H-Indol-2-one, 1,3-dihydro-3-[(4-methylphenyl)methylene]-, (3E)-](http://img.cochemist.com/ccimg/90900/90828-09-4.png)
![2H-Indol-2-one, 1,3-dihydro-3-[(4-methylphenyl)methylene]-, (3E)-](http://img.cochemist.com/ccimg/90900/90828-09-4_b.png)
![Phenol, 2,2'-[1,2-ethanediylbis(iminoethylidene)]bis-](http://img.cochemist.com/ccimg/92000/91915-31-0.png)
![Phenol, 2,2'-[1,2-ethanediylbis(iminoethylidene)]bis-](http://img.cochemist.com/ccimg/92000/91915-31-0_b.png)
![2-Pyridinamine, N-3H-[1,2,4]thiadiazolo[4,3-a]pyridin-3-ylidene-](http://img.cochemist.com/ccimg/24100/24097-57-2.png)
![2-Pyridinamine, N-3H-[1,2,4]thiadiazolo[4,3-a]pyridin-3-ylidene-](http://img.cochemist.com/ccimg/24100/24097-57-2_b.png)
![2H-Indol-2-one, 3-[(4-chlorophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-52-8.png)
![2H-Indol-2-one, 3-[(4-chlorophenyl)methylene]-1,3-dihydro-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-52-8_b.png)









![2H-Indol-2-one, 1,3-dihydro-3-[(4-methoxyphenyl)methylene]-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-48-2.png)
![2H-Indol-2-one, 1,3-dihydro-3-[(4-methoxyphenyl)methylene]-, (3E)-](http://img.cochemist.com/ccimg/29600/29551-48-2_b.png)




![BORANE, HYDROXYBIS[(TRIPHENYLSTANNYL)OXY]-, COMPD. WITH METHYLBENZENE(1:1)](http://img.cochemist.com/ccimg/404600/404595-91-1.png)
![BORANE, HYDROXYBIS[(TRIPHENYLSTANNYL)OXY]-, COMPD. WITH METHYLBENZENE(1:1)](http://img.cochemist.com/ccimg/404600/404595-91-1_b.png)








![2-PENTANONE, 5-[1-(DIMETHYLPHENYLSILYL)-2-METHYLENECYCLOPROPYL]-](http://img.cochemist.com/ccimg/641700/641614-27-9.png)
![2-PENTANONE, 5-[1-(DIMETHYLPHENYLSILYL)-2-METHYLENECYCLOPROPYL]-](http://img.cochemist.com/ccimg/641700/641614-27-9_b.png)














![Benzonitrile, 4-[(1E)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/95800/95759-33-4.png)
![Benzonitrile, 4-[(1E)-3-oxo-1-propenyl]-](http://img.cochemist.com/ccimg/95800/95759-33-4_b.png)





![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)


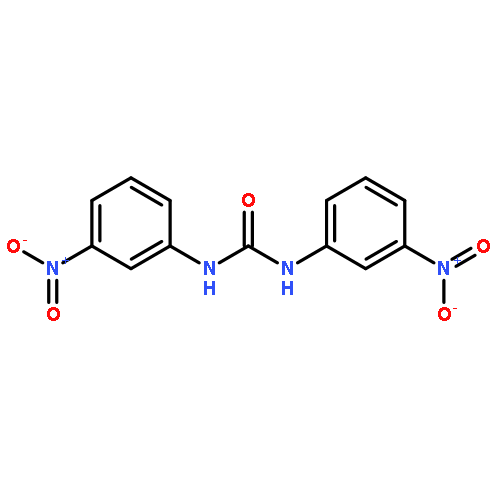
![Urea, N,N'-bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2000/1960-88-9.png)
![Urea, N,N'-bis[4-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/2000/1960-88-9_b.png)
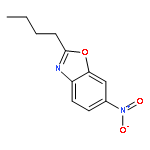









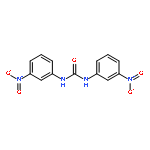






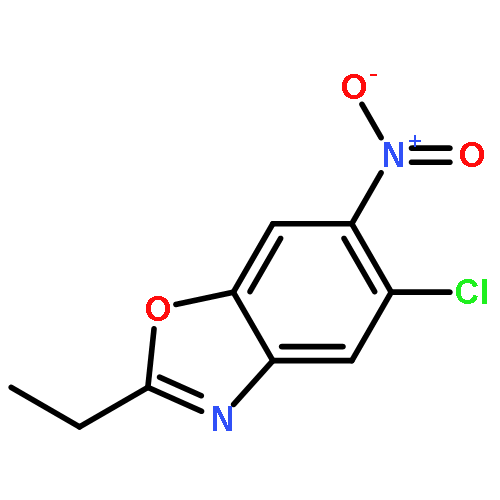
![2-(Chloromethyl)-6-nitrobenzo[d]oxazole](http://img.cochemist.com/ccimg/221700/221638-74-0.png)
![2-(Chloromethyl)-6-nitrobenzo[d]oxazole](http://img.cochemist.com/ccimg/221700/221638-74-0_b.png)



![1H-Inden-1-one, 6-[2-(b-D-glucopyranosyloxy)ethyl]-2,3-dihydro-3-hydroxy-7-(hydroxymethyl)-2,5-dimethyl-,(2S,3S)-](http://img.cochemist.com/ccimg/62100/62043-50-9.png)
![1H-Inden-1-one, 6-[2-(b-D-glucopyranosyloxy)ethyl]-2,3-dihydro-3-hydroxy-7-(hydroxymethyl)-2,5-dimethyl-,(2S,3S)-](http://img.cochemist.com/ccimg/62100/62043-50-9_b.png)




















![2-ethyl-Oxazolo[5,4-b]pyridine](http://img.cochemist.com/ccimg/857000/856990-30-2.png)
![2-ethyl-Oxazolo[5,4-b]pyridine](http://img.cochemist.com/ccimg/857000/856990-30-2_b.png)









![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6.png)
![1,3-BIS[3,5-BIS(TRIFLUOROMETHYL)PHENYL]UREA](http://img.cochemist.com/ccimg/3900/3824-74-6_b.png)









![bicyclo[2.2.1]hept-5-ene-2-carbonyl chloride](http://img.cochemist.com/ccimg/27100/27063-48-5.png)
![bicyclo[2.2.1]hept-5-ene-2-carbonyl chloride](http://img.cochemist.com/ccimg/27100/27063-48-5_b.png)

![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-methyl-α-methylene-, methyl ester](/data/chemimg/167300/1187847-13-7.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-methyl-α-methylene-, methyl ester](/data/chemimg/167300/1187847-13-7_b.png)
![Benzenepropanoic acid, 4-chloro-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1132641-29-2.png)
![Benzenepropanoic acid, 4-chloro-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1132641-29-2_b.png)
![6-[(9H-fluoren-2-ylamino)methylidene]cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/14000/13924-55-5.png)
![6-[(9H-fluoren-2-ylamino)methylidene]cyclohexa-2,4-dien-1-one](http://img.cochemist.com/ccimg/14000/13924-55-5_b.png)



![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-fluoro-α-methylene-, methyl ester](/data/chemimg/141200/1132641-31-6.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-4-fluoro-α-methylene-, methyl ester](/data/chemimg/141200/1132641-31-6_b.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)


![Benzenepropanoic acid, 2-bromo-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1202888-62-7.png)
![Benzenepropanoic acid, 2-bromo-β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/141200/1202888-62-7_b.png)







![Acetic acid, 2,2'-[(11,23-dihydroxypentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-5,17-diyl)bis(oxy)]bis-, 1,1'-diethyl ester](/data/chemimg/110600/142267-55-8.png)
![Acetic acid, 2,2'-[(11,23-dihydroxypentacyclo[19.3.1.13,7.19,13.115,19]octacosa-1(25),3,5,7(28),9,11,13(27),15,17,19(26),21,23-dodecaene-5,17-diyl)bis(oxy)]bis-, 1,1'-diethyl ester](/data/chemimg/110600/142267-55-8_b.png)


![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0.png)
![1,3-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea](/data/chemimg/315700/1060-92-0_b.png)






![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/105500/956833-12-8.png)
![Benzenepropanoic acid, β-[[(1,1-dimethylethoxy)carbonyl]oxy]-α-methylene-, methyl ester](/data/chemimg/105500/956833-12-8_b.png)