Co-reporter:Shan Tang;Lu-Jun Liang;Yan-Yan Si;Shuai Gao;Jia-Xing Wang;Jun Liang; Ziqing Mei; Ji-Shen Zheng; Lei Liu
Angewandte Chemie 2017 Volume 129(Issue 43) pp:13518-13522
Publication Date(Web):2017/10/16
DOI:10.1002/ange.201708067
AbstractChemical ubiquitination is an effective approach for accessing structurally defined, atypical ubiquitin (Ub) chains that are difficult to prepare by other techniques. Herein, we describe a strategy that uses a readily accessible premade isopeptide-linked 76-mer (isoUb), which has an N-terminal Cys and a C-terminal hydrazide, as the key building block to assemble atypical Ub chains in a modular fashion. This method avoids the use of auxiliary-modified Lys and instead employs the canonical and therefore more robust Cys-based native chemical ligation technique. The efficiency and capacity of this isoUb-based strategy is exemplified by the cost-effective synthesis of several linkage- and length-defined atypical Ub chains, including K27-linked tetra-Ub and K11/K48-branched tri-, tetra-, penta-, and hexa-Ubs.
Co-reporter:Qiaoqiao He;Jiabin Li;Yunkun Qi;Zhipeng Wang;Yong Huang
Science China Chemistry 2017 Volume 60( Issue 5) pp:621-627
Publication Date(Web):14 December 2016
DOI:10.1007/s11426-016-0386-4
Histone H2A methylation at Gln104 (H2AQ104Me) is a new type of histone post-translational modification (PTM) discovered recently. This modification has been found to have significant influence on gene transcription. However, the structural and functional consequence of glutamine methylation on nucleosome remains to be further elucidated. Obtaining of histones with site-specific methylation at glutamine residues might facilitate the studies towards a better understanding of this new PTM. In the present work, total chemical synthesis of H2AQ104Me was carried out through use of the hydrazide-based native chemical ligation. Synthetic histone H2AQ104Me could be successfully incorporated into nucleosomes in vitro and showed a negative influence on the nucleosome stability.
Co-reporter:Bingjia Yan, Linzhi Ye, Weiliang Xu, Lei Liu
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 18(Issue 18) pp:
Publication Date(Web):15 September 2017
DOI:10.1016/j.bmc.2017.05.020
Solution of the three-dimensional structures of proteins is a critical step in deciphering the molecular mechanisms of their bioactivities. Among the many approaches for obtaining protein crystals, racemic protein crystallography has been developed as a unique method to solve the structures of an increasing number of proteins. Exploiting unnatural protein enantiomers in crystallization and resolution, racemic protein crystallography manifests two major advantages that are 1) to increase the success rate of protein crystallization, and 2) to obviate the phase problem in X-ray diffraction. The requirement of unnatural protein enantiomers in racemic protein crystallography necessitates chemical protein synthesis, which is hitherto accomplished through solid phase peptide synthesis and chemical ligation reactions. This review highlights the fundamental ideas of racemic protein crystallography and surveys the harvests in the field of racemic protein crystallography over the last five years from early 2012 to late 2016.Download high-res image (175KB)Download full-size image
Co-reporter:Man Pan; Shuai Gao; Yong Zheng; Xiaodan Tan; Huan Lan; Xianglong Tan; Demeng Sun; Lining Lu; Tian Wang; Qingyun Zheng; Yichao Huang; Jiawei Wang
Journal of the American Chemical Society 2016 Volume 138(Issue 23) pp:7429-7435
Publication Date(Web):June 6, 2016
DOI:10.1021/jacs.6b04031
Quasi-racemic crystallography has been used to determine the X-ray structures of K27-linked ubiquitin (Ub) chains prepared through total chemical synthesis. Crystal structures of K27-linked di- and tri-ubiquitins reveal that the isopeptide linkages are confined in a unique buried conformation, which provides the molecular basis for the distinctive function of K27 linkage compared to the other seven Ub chains. K27-linked di- and triUb were found to adopt different structural conformations in the crystals, one being symmetric whereas the other triangular. Furthermore, bioactivity experiments showed that the ovarian tumor family de-ubiquitinase 2 significantly favors K27-linked triUb than K27-linked diUb. K27-linked triUb represents the so-far largest chemically synthesized protein (228 amino acids) that has been crystallized to afford a high-resolution X-ray structure.
Co-reporter:Shuai Gao, Man Pan, Yong Zheng, Yichao Huang, Qingyun Zheng, Demeng Sun, Lining Lu, Xiaodan Tan, Xianglong Tan, Huan Lan, Jiaxing Wang, Tian Wang, Jiawei Wang, and Lei Liu
Journal of the American Chemical Society 2016 Volume 138(Issue 43) pp:14497-14502
Publication Date(Web):October 21, 2016
DOI:10.1021/jacs.6b09545
Racemic or quasi-racemic crystallography recently emerges as a useful technology for solution of the crystal structures of biomacromolecules. It remains unclear to what extent the biomacromolecules of opposite handedness can differ from each other in racemic or quasi-racemic crystallography. Here we report a finding that monomeric d-ubiquitin (Ub) has propensity to cocrystallize with different dimers, trimers, and even a tetramer of l-Ub. In these cocrystals the unconnected monomeric d-Ubs can self-assemble to form pseudomirror images of different oligomers of l-Ub. This monomer/oligomer cocrystallization phenomenon expands the concept of racemic crystallography. Using the monomer/oligomer cocrystallization technology we obtained, for the first time the X-ray structures of linear M1-linked tri- and tetra-Ubs and a K11/K63-branched tri-Ub.
Co-reporter:Shan Tang, Zhengpeng Wan, Yiren Gao, Ji-Shen Zheng, Jing Wang, Yan-Yan Si, Xin Chen, Hai Qi, Lei Liu and Wanli Liu
Chemical Science 2016 vol. 7(Issue 3) pp:1891-1895
Publication Date(Web):11 Dec 2015
DOI:10.1039/C5SC03404C
We report the chemical synthesis of the first photo-activatable protein antigen that can be used to study antigen–antibody interaction mediated responses in B cells. This strategy facilitated fine tuning of the caged protein antigen to optimize its bioactivity and photochemical properties. One optimal molecule, HEL-K96NPE, was totally inert to hen egg lysozyme (HEL)-specific B cells and could only restore its antigenicity upon photoactivation. Combined with real time live cell imaging, the utility of HEL-K96NPE was demonstrated as a proof of concept to quantify B cell synapse formation and calcium influx responses at the single cell level.
Co-reporter:Yun-Kun Qi, Shan Tang, Yi-Chao Huang, Man Pan, Ji-Shen Zheng and Lei Liu
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 18) pp:4194-4198
Publication Date(Web):14 Apr 2016
DOI:10.1039/C6OB00450D
A new thiol protecting group Hmboff/on is described, which has a switchable activity that may be useful in the chemical synthesis of proteins. When placed on the side chain of Cys, Cys(Hmboff) is stable to trifluoroacetic acid (TFA) in the process of solid-phase peptide synthesis. When Cys(Hmboff) is treated with neutral aqueous buffers, it is cleanly converted to acid-labile Cys(Hmbon), which can later be fully deprotected by TFA to generate free Cys. The utility of Cys(Hmboff/on) is demonstrated by the chemical synthesis of an erythropoietin segment, EPO[Cys98–Arg166]-OH through native chemical ligation.
Co-reporter:Xi Lu, Bin Xiao, Rui Shang, Lei Liu
Chinese Chemical Letters 2016 Volume 27(Issue 3) pp:305-311
Publication Date(Web):March 2016
DOI:10.1016/j.cclet.2015.12.021
Unnatural α-amino acids have been extensively used in the modern drug discovery and protein engineering studies. They have also found applications in the development of chiral molecular catalysts and the total synthesis of diverse natural products. Accordingly the development of cost-effective approaches for the preparation of unnatural α-amino acids has received increasing attentions. Among all the available methods for this purpose, direct CH functionalization of simple amino acids represents one of the most attractive approaches because it exhibits good atom-economy and step-efficiency. In particular, selective functionalization of either the primary or secondary C(sp3)H bonds in the amino acids has been explored to make versatile CC, CN, CO, CB and CF bonds to modify the side chain of amino acids and even peptides. The present review surveys the recent advances of synthesis of chiral unnatural α-amino acids and peptides through palladium-catalyzed functionalization of un-activated C(sp3)H bonds.Recent advances in the synthesis of chiral unnatural amino acids and peptides through palladium-catalyzed C(sp3)H functionalization are discussed.
Co-reporter:Jing Wang;Yiyun Cao;Yiren Gao;Zhengpeng Wan;Shan Tang;Junyang Yi;Yanyan Si;Haowen Zhang;Wanli Liu
PNAS 2016 Volume 113 (Issue 5 ) pp:E558-E567
Publication Date(Web):2016-02-02
DOI:10.1073/pnas.1517612113
Antigen binding to the B-cell receptor (BCR) induces several responses, resulting in B-cell activation, proliferation, and
differentiation. However, it has been difficult to study these responses due to their dynamic, fast, and transient nature.
Here, we attempted to solve this problem by developing a controllable trigger point for BCR and antigen recognition through
the construction of a photoactivatable antigen, caged 4-hydroxy-3-nitrophenyl acetyl (caged-NP). This photoactivatable antigen
system in combination with live cell and single molecule imaging techniques enabled us to illuminate the previously unidentified
B-cell probing termination behaviors and the precise BCR sorting mechanisms during B-cell activation. B cells in contact with
caged-NP exhibited probing behaviors as defined by the unceasing extension of membrane pseudopods in random directions. Further
analyses showed that such probing behaviors are cell intrinsic with strict dependence on F-actin remodeling but not on tonic
BCR signaling. B-cell probing behaviors were terminated within 4 s after photoactivation, suggesting that this response was
sensitive and specific to BCR engagement. The termination of B-cell probing was concomitant with the accumulation response
of the BCRs into the BCR microclusters. We also determined the Brownian diffusion coefficient of BCRs from the same B cells
before and after BCR engagement. The analysis of temporally segregated single molecule images of both BCR and major histocompatibility
complex class I (MHC-I) demonstrated that antigen binding induced trapping of BCRs into the BCR microclusters is a fundamental
mechanism for B cells to acquire antigens.
Co-reporter:Chunlin Lv;Hao Cheng;Wei He;Muhammad Ishaq Ali Shah;Congqiao Xu
Nano Research 2016 Volume 9( Issue 9) pp:2544-2550
Publication Date(Web):2016 September
DOI:10.1007/s12274-016-1140-8
Identification of metal cluster catalysis is a topic that is being investigated since a long time. Here, we report a Pd3 metal cluster catalytic reaction investigated by means of operando studies. We discovered that atomically defined tri-nuclear palladium (Pd3) is a surprisingly active catalyst for the cycloisomerization of 2-phenylethynylaniline. Operando 1H NMR spectroscopy and X-ray extended absorption fine structure (EXAFS) measurements have indicated that the structural integrity of such a catalyst remains intact throughout the reaction, which has also been confirmed by an ex situ X-ray photoelectron spectroscopy (XPS) study and catalyst recycling experiments. Kinetic data derived from operando IR spectroscopy measurements have shown that Pd3 is the active catalytic species. Density functional theory calculations have revealed a reaction pathway consistent with the kinetic data, further supported by NMR titration and X-ray crystal structure studies. Overall, the present study presents a clear example of metal cluster catalysis.
Co-reporter:Bin Xiao; Zhiqiang Niu; Yang-Gang Wang; Wei Jia; Jian Shang; Lan Zhang; Dingsheng Wang; Yao Fu; Jie Zeng; Wei He; Kai Wu; Jun Li; Jinlong Yang; Lei Liu;Yadong Li
Journal of the American Chemical Society 2015 Volume 137(Issue 11) pp:3791-3794
Publication Date(Web):March 17, 2015
DOI:10.1021/jacs.5b01391
Previous studies have shown that crystal planes of heterogeneous catalysts could display enhanced activity, such that higher turnover or chemoselectivity could be achieved. Here we report an example where the reaction stereoselectivity was significantly affected by the catalyst crystal planes. In copper-catalyzed deoxygenation reaction of aromatic epoxides, copper cubes, wires, and plates gave the olefin products with different cis/trans selectivities, whereas homogeneous copper catalysts showed poor selectivity. Scanning tunneling microscope and density functional theory studies revealed that the different adsorption mode and higher adsorption strength of epoxide oxygen on Cu{100} plane were responsible for the observed variation of selectivity. The copper-catalyzed deoxygenation reaction provided new practical access to cis-olefins from readily available aromatic epoxides. Our work also indicated that nanocrystal catalysts may provide useful stereochemical control in organic reactions.
Co-reporter:Wenzhong Yan, Jie Qing, Hanbing Mei, Junxiu Nong, Jin Huang, Jin Zhu, Hualiang Jiang, Lei Liu, Linqi Zhang, Jian Li
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 24) pp:5682-5686
Publication Date(Web):15 December 2015
DOI:10.1016/j.bmcl.2015.11.002
In this work, the relationship between cyclophilin A (CypA) and EV71 prompted us to screen a series of small molecular CypA inhibitors which were previously reported by our group. Among them, compounds 1 and 2 were discovered as non-immunosuppressive anti-EV71 agents with an EC50 values of 1.07 ± 0.17 μM and 3.36 ± 0.45 μM in virus assay, respectively, which were desirably for the further study. The subsequent chemical modifications derived a novel class of molecules, among which compound 11 demonstrated the most potent anti-EV71 activity in virus assay (EC50 = 0.37 ± 0.17 μM), and low cytotoxicity (CC50 > 25 μM). The following CypA enzyme inhibition studies indicated that there was not only the enzyme inhibition activity, undoubtedly important, functioning in the antiviral process, but also some unknown mechanisms worked in combination, and the further study is underway in our laboratory. Nevertheless, to the best of our knowledge, compound 11 was probably the most potent small molecular anti-EV71 agent via CypA inhibitory mechanism to date. Consequently, our study provided a new potential small molecule for curing EV71 infection.In this work, the relationship between cyclophilin A (CypA) and EV71 prompted us to screen a series of small molecular CypA inhibitors which were previously reported by our group. Among them, compounds 1 and 2 were discovered as non-immunosuppressive anti-EV71 agents with an EC50 values of 1.07 ± 0.17 μM and 3.36 ± 0.45 μM in virus assay, respectively, which were desirably for the further study. The subsequent chemical modifications derived a novel class of molecules, and the structure–activity relationship of these derivatives in the virus assays demonstrated that the 2,6-dihydroxy-benzoylurea moiety and the planar fluorene ring were crucial for the anti-EV71 ability. Furthermore, the results of the modification on the fluorene ring indicated that the substitution at 3′ site of the fluorene ring was better than 1′ site and fluorine was the best substituent. Eventually, among these compounds, 11 demonstrated the most potent anti-EV71 activity in virus assay (EC50 = 0.37 ± 0.17 μM), and low cytotoxicity (CC50 > 25 μM). The following CypA enzyme inhibition studies indicated that there was not only the enzyme inhibition activity, undoubtedly important, functioning in the antiviral process, but also some unknown mechanisms worked in combination, and the further study is underway in our laboratory. Nevertheless, to the best of our knowledge, compound 11 was probably the most potent small molecular anti-EV71 agent via CypA inhibitory mechanism to date. Consequently, our study provided a new potential small molecule for curing EV71 infection.
Co-reporter:Hao-Nan Chang;Bei-Yuan Liu;Yun-Kun Qi;Yang Zhou;Yan-Ping Chen;Kai-Mai Pan;Wen-Wen Li;Xiu-Man Zhou;Wei-Wei Ma; Cai-Yun Fu; Yuan-Ming Qi; Lei Liu; Yan-Feng Gao
Angewandte Chemie International Edition 2015 Volume 54( Issue 40) pp:
Publication Date(Web):
DOI:10.1002/anie.201506225
Abstract
Blockade of the protein–protein interaction between the transmembrane protein programmed cell death protein 1 (PD-1) and its ligand PD-L1 has emerged as a promising immunotherapy for treating cancers. Using the technology of mirror-image phage display, we developed the first hydrolysis-resistant D-peptide antagonists to target the PD-1/PD-L1 pathway. The optimized compound DPPA-1 could bind PD-L1 at an affinity of 0.51 μM in vitro. A blockade assay at the cellular level and tumor-bearing mice experiments indicated that DPPA-1 could also effectively disrupt the PD-1/PD-L1 interaction in vivo. Thus D-peptide antagonists may provide novel low-molecular-weight drug candidates for cancer immunotherapy.
Co-reporter:Jia-Xing Wang;Ge-Min Fang;Yao He;Da-Liang Qu;Min Yu; Zhang-Yong Hong;Dr. Lei Liu
Angewandte Chemie International Edition 2015 Volume 54( Issue 7) pp:2194-2198
Publication Date(Web):
DOI:10.1002/anie.201408078
Abstract
Fully unprotected peptide o-aminoanilides can be efficiently activated by NaNO2 in aqueous solution to furnish peptide thioesters for use in native chemical ligation. This finding enables the convergent synthesis of proteins from readily synthesizable peptide o-aminoanilides as a new type of crypto-thioesters. The practicality of this approach is shown by the synthesis of histone H2B from five peptide segments. Purification or solubilization tags, which are sometimes needed to improve the efficiency of protein chemical synthesis, can be incorporated into the o-aminoanilide moiety, as demonstrated in the preparation of the cyclic protein lactocyclicin Q.
Co-reporter:Yichao Huang
Science China Chemistry 2015 Volume 58( Issue 12) pp:1779-1781
Publication Date(Web):2015 December
DOI:10.1007/s11426-015-5462-2
Co-reporter:Shan Tang;Yan-Yan Si;Zhi-Peng Wang;Kun-Rong Mei;Xin Chen;Jing-Yuan Cheng;Dr. Ji-Shen Zheng;Dr. Lei Liu
Angewandte Chemie International Edition 2015 Volume 54( Issue 19) pp:5713-5717
Publication Date(Web):
DOI:10.1002/anie.201500051
Abstract
Successive peptide ligation using a one-pot method can improve the efficiency of protein chemical synthesis. Although one-pot three-segment ligation has enjoyed widespread application, a robust method for one-pot four-segment ligation had to date remained undeveloped. Herein we report a new one-pot multisegment peptide ligation method that can be used to condense up to four segments with operational simplicity and high efficiency. Its practicality is demonstrated by the one-pot four-segment synthesis of a plant protein, crambin, and a human chemokine, hCCL21.
Co-reporter:Yuanye Jiang;Haizhu Yu;Yao Fu
Science China Chemistry 2015 Volume 58( Issue 4) pp:673-683
Publication Date(Web):2015 April
DOI:10.1007/s11426-014-5178-8
Trifluoromethylation reactions are important transformations in the research and development of drugs, agrochemicals and functional materials. An oxidation/reduction process of trifluoromethyl-containing compounds is thought to be involved in many recently tested catalytic trifluoromethylation reactions. To provide helpful physical chemical data for mechanistic studies on trifluoromethylation reactions, the redox potentials of a variety of trifluoromethyl-containing compounds and trifluoromethylated radicals were studied by quantum-chemical methods. First, wB97X-D was found to be a reliable method in predicting the ionization potentials, electron affinities, bond dissociation enthalpies and redox potentials of trifluoromethyl-containing compounds. One-electron absolute redox potentials of 79 trifluoromethyl substrates and 107 trifluoromethylated radicals in acetonitrile were then calculated with this method. The theoretical results were found to be helpful for interpreting experimental observations such as the relative reaction efficiency of different trifluoromethylation reagents. Finally, the bond dissociation free energies (BDFE) of various compounds were found to have a good linear relationship with the related bond dissociation enthalpies (BDE). Based on this observation, a convenient method was proposed to predict one-electron redox potentials of neutral molecules.
Co-reporter:Hao-Nan Chang;Bei-Yuan Liu;Yun-Kun Qi;Yang Zhou;Yan-Ping Chen;Kai-Mai Pan;Wen-Wen Li;Xiu-Man Zhou;Wei-Wei Ma; Cai-Yun Fu; Yuan-Ming Qi; Lei Liu; Yan-Feng Gao
Angewandte Chemie 2015 Volume 127( Issue 40) pp:
Publication Date(Web):
DOI:10.1002/ange.201506225
Abstract
Blockade of the protein–protein interaction between the transmembrane protein programmed cell death protein 1 (PD-1) and its ligand PD-L1 has emerged as a promising immunotherapy for treating cancers. Using the technology of mirror-image phage display, we developed the first hydrolysis-resistant D-peptide antagonists to target the PD-1/PD-L1 pathway. The optimized compound DPPA-1 could bind PD-L1 at an affinity of 0.51 μM in vitro. A blockade assay at the cellular level and tumor-bearing mice experiments indicated that DPPA-1 could also effectively disrupt the PD-1/PD-L1 interaction in vivo. Thus D-peptide antagonists may provide novel low-molecular-weight drug candidates for cancer immunotherapy.
Co-reporter:Ye Guo;De-Meng Sun;Feng-Liang Wang;Yao He; Lei Liu; Chang-Lin Tian
Angewandte Chemie 2015 Volume 127( Issue 48) pp:14484-14489
Publication Date(Web):
DOI:10.1002/ange.201500699
Abstract
Disulfide-rich peptides containing three or more disulfide bonds are promising therapeutic and diagnostic agents, but their preparation is often limited by the tedious and low-yielding folding process. We found that a single cystine-to-diaminodiacid replacement could significantly increase the folding efficiency of disulfide-rich peptides and thus improve their production yields. The practicality of this strategy was demonstrated by the synthesis and folding of derivatives of the μ-conotoxin SIIIA, the preclinical hormone hepcidin, and the trypsin inhibitor EETI-II. NMR and X-ray crystallography studies confirmed that these derivatives of disulfide-rich peptide retained the correct three-dimensional conformations. Moreover, the cystine-to-diaminodiacid replacement enabled structural tuning, thereby leading to an EETI-II derivative with higher bioactivity than the native peptide.
Co-reporter:Ye Guo;De-Meng Sun;Feng-Liang Wang;Yao He; Lei Liu; Chang-Lin Tian
Angewandte Chemie International Edition 2015 Volume 54( Issue 48) pp:14276-14281
Publication Date(Web):
DOI:10.1002/anie.201500699
Abstract
Disulfide-rich peptides containing three or more disulfide bonds are promising therapeutic and diagnostic agents, but their preparation is often limited by the tedious and low-yielding folding process. We found that a single cystine-to-diaminodiacid replacement could significantly increase the folding efficiency of disulfide-rich peptides and thus improve their production yields. The practicality of this strategy was demonstrated by the synthesis and folding of derivatives of the μ-conotoxin SIIIA, the preclinical hormone hepcidin, and the trypsin inhibitor EETI-II. NMR and X-ray crystallography studies confirmed that these derivatives of disulfide-rich peptide retained the correct three-dimensional conformations. Moreover, the cystine-to-diaminodiacid replacement enabled structural tuning, thereby leading to an EETI-II derivative with higher bioactivity than the native peptide.
Co-reporter:Jia-Xing Wang;Ge-Min Fang;Yao He;Da-Liang Qu;Min Yu; Zhang-Yong Hong;Dr. Lei Liu
Angewandte Chemie 2015 Volume 127( Issue 7) pp:2222-2226
Publication Date(Web):
DOI:10.1002/ange.201408078
Abstract
Fully unprotected peptide o-aminoanilides can be efficiently activated by NaNO2 in aqueous solution to furnish peptide thioesters for use in native chemical ligation. This finding enables the convergent synthesis of proteins from readily synthesizable peptide o-aminoanilides as a new type of crypto-thioesters. The practicality of this approach is shown by the synthesis of histone H2B from five peptide segments. Purification or solubilization tags, which are sometimes needed to improve the efficiency of protein chemical synthesis, can be incorporated into the o-aminoanilide moiety, as demonstrated in the preparation of the cyclic protein lactocyclicin Q.
Co-reporter:Shan Tang;Yan-Yan Si;Zhi-Peng Wang;Kun-Rong Mei;Xin Chen;Jing-Yuan Cheng;Dr. Ji-Shen Zheng;Dr. Lei Liu
Angewandte Chemie 2015 Volume 127( Issue 19) pp:5805-5809
Publication Date(Web):
DOI:10.1002/ange.201500051
Abstract
Successive peptide ligation using a one-pot method can improve the efficiency of protein chemical synthesis. Although one-pot three-segment ligation has enjoyed widespread application, a robust method for one-pot four-segment ligation had to date remained undeveloped. Herein we report a new one-pot multisegment peptide ligation method that can be used to condense up to four segments with operational simplicity and high efficiency. Its practicality is demonstrated by the one-pot four-segment synthesis of a plant protein, crambin, and a human chemokine, hCCL21.
Co-reporter:Ji-Shen Zheng ; Mu Yu ; Yun-Kun Qi ; Shan Tang ; Fei Shen ; Zhi-Peng Wang ; Liang Xiao ; Longhua Zhang ; Chang-Lin Tian
Journal of the American Chemical Society 2014 Volume 136(Issue 9) pp:3695-3704
Publication Date(Web):February 21, 2014
DOI:10.1021/ja500222u
Total chemical synthesis provides a unique approach for the access to uncontaminated, monodisperse, and more importantly, post-translationally modified membrane proteins. In the present study we report a practical procedure for expedient and cost-effective synthesis of small to medium-sized membrane proteins in multimilligram scale through the use of automated Fmoc chemistry. The key finding of our study is that after the attachment of a removable arginine-tagged backbone modification group, the membrane protein segments behave almost the same as ordinary water-soluble peptides in terms of Fmoc solid-phase synthesis, ligation, purification, and mass spectrometry characterization. The efficiency and practicality of the new method is demonstrated by the successful preparation of Ser64-phosphorylated M2 proton channel from influenza A virus and the membrane-embedded domain of an inward rectifier K+ channel protein Kir5.1. Functional characterizations of these chemically synthesized membrane proteins indicate that they provide useful and otherwise-difficult-to-access materials for biochemistry and biophysics studies.
Co-reporter:Jiabin Li, Yuanyuan Li, Qiaoqiao He, Yiming Li, Haitao Li and Lei Liu
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 29) pp:5435-5441
Publication Date(Web):17 Jun 2014
DOI:10.1039/C4OB00715H
One of the rising demands in the field of protein chemical synthesis is the development of facile strategies that yield the protein in workable quantities and homogeneity, with fewer handling steps. Although the native chemical ligation of peptide hydrazides has recently been shown to be useful for the chemical synthesis of proteins carrying acid-sensitive modification groups, previous hydrazide-based protein synthesis studies have used sequential ligation strategies. Here, we report a practical method for a “one-pot” native chemical ligation of peptide hydrazides that would circumvent the need for the isolation of the intermediate products. This method employed a fast and selective arylboronate oxidation reaction mediated by H2O2, which draws attention to the potential applications of the thus far under-exploited boron-based functionalities in protein chemical synthesis. To demonstrate the practicality and efficiency of the new one-pot method, we report its application to a scalable total synthesis of modified histones (with five analogues of H3 and H4 as examples) on a multi-milligram scale, with good homogeneity.
Co-reporter:Xi Lu;Jun Yi;Zhen-Qi Zhang;Jian-Jun Dai;Jing-Hui Liu; Bin Xiao; Yao Fu; Lei Liu
Chemistry - A European Journal 2014 Volume 20( Issue 47) pp:15339-15343
Publication Date(Web):
DOI:10.1002/chem.201405296
Abstract
A novel method for the synthesis of non-natural L- and D-amino acids by a Ni-catalyzed reductive cross-coupling reaction is described. This strategy enables the racemization-free cross-coupling of serine/homoserine- derived iodides with aryl/acyl/alkyl halides. It provides convenient access to varieties of enantiopure and functionalized amino acids, which are important building blocks in bioactive compounds and pharmaceuticals.
Co-reporter:Jing-Hui Liu;Chu-Ting Yang;Xiao-Yu Lu;Zhen-Qi Zhang;Ling Xu;Mian Cui;Xi Lu;Bin Xiao;Dr. Yao Fu;Dr. Lei Liu
Chemistry - A European Journal 2014 Volume 20( Issue 47) pp:15334-15338
Publication Date(Web):
DOI:10.1002/chem.201405223
Abstract
A copper-catalyzed reductive cross-coupling reaction of nonactivated alkyl tosylates and mesylates with alkyl and aryl bromides was developed. It provides a practical method for efficient and cost-effective construction of aryl–alkyl and alkyl–alkyl CC bonds with stereocontrol from readily available substrates. When used in an intramolecular fashion, the reaction enables convenient access to various substituted carbo- or heterocycles, such as 2,3-dihydrobenzofuran and benzochromene derivatives.
Co-reporter:Lei Liu
Tetrahedron 2014 70(42) pp: 7620
Publication Date(Web):
DOI:10.1016/j.tet.2014.08.006
Co-reporter:Lei Liu
Tetrahedron 2014 70(42) pp: 7619
Publication Date(Web):
DOI:10.1016/j.tet.2014.08.007
Co-reporter:Yi-Ming Li;Yi-Tong Li;Man Pan;Xiu-Qi Kong;Yi-Chao Huang; Zhang-Yong Hong; Lei Liu
Angewandte Chemie 2014 Volume 126( Issue 8) pp:2230-2234
Publication Date(Web):
DOI:10.1002/ange.201310010
Abstract
Sortase-mediated hydrazinolysis of proteins with hydrazine or its derivatives was developed for the production of recombinant protein hydrazides. This process provides an alternative approach for protein semisynthesis through the use of recombinant protein hydrazides as thioester surrogates. It also provides an alternative method for C-terminal modification of proteins with functional units as well as for the preparation of C-to-C fusion proteins.
Co-reporter:Man Pan;Shan Li;Xiang Li;Dr. Feng Shao;Dr. Lei Liu;Dr. Hong-Gang Hu
Angewandte Chemie International Edition 2014 Volume 53( Issue 52) pp:14517-14521
Publication Date(Web):
DOI:10.1002/anie.201407824
Abstract
As a unique and unappreciated protein posttranslational modification, arginine N-glycosylation was recently discovered to play an important role in the process that bacteria counteract host defenses. To provide chemical tools for further proteomic and biochemical studies on arginine N-glycosylation, we report the first general strategy for a rapid and cost-effective synthesis of glycopeptides carrying single or multiple arginine N-GlcNAcyl groups. These glycopeptides were successfully utilized to generate the first antibodies that can specifically recognize arginine N-GlcNAcylated peptides or proteins in a sequence-independent manner.
Co-reporter:Yi-Ming Li;Yi-Tong Li;Man Pan;Xiu-Qi Kong;Yi-Chao Huang; Zhang-Yong Hong; Lei Liu
Angewandte Chemie International Edition 2014 Volume 53( Issue 8) pp:2198-2202
Publication Date(Web):
DOI:10.1002/anie.201310010
Abstract
Sortase-mediated hydrazinolysis of proteins with hydrazine or its derivatives was developed for the production of recombinant protein hydrazides. This process provides an alternative approach for protein semisynthesis through the use of recombinant protein hydrazides as thioester surrogates. It also provides an alternative method for C-terminal modification of proteins with functional units as well as for the preparation of C-to-C fusion proteins.
Co-reporter:Hong-Kui Cui, Bing Zhao, Yehua Li, Ye Guo, Hao Hu, Lei Liu and Ye-Guang Chen
Cell Research 2013 23(4) pp:581-584
Publication Date(Web):February 26, 2013
DOI:10.1038/cr.2013.30
The canonical Wnt/β-catenin signaling pathway orchestrates cell morphology, motility, proliferation and differentiation. This pathway plays important roles in embryogenesis, adult tissue homeostasis and tissue regeneration. Aberrant activation of the Wnt/β-catenin signaling pathway has been implicated in the development of a broad spectrum of tumors, while attenuation of this pathway contributes to a number of human diseases including osteoporosis, neurodegenerative diseases, diabetes and Joubert syndrome1. The design of organic compounds that modulate Wnt/β-catenin signaling constitutes an interesting strategy for therapeutic intervention of this key pathway. Additionally, as the Wnt/β-catenin signaling pathway is critical for the maintenance of embryonic stem cells and multiple types of adult stem cells, agonists or antagonists of Wnt/β-catenin signaling may provide useful tools for the studies of stem cell self-renewal and differentiation, and tissue regeneration.The design of hydrocarbon-stapled peptides was developed by Verdine et al.2 to mimic the α-helix conformation of folded proteins. This method has been demonstrated to efficiently increase the helical propensity and the binding ability of peptides. In vivo, stapled peptides can penetrate through cell membranes to attack the intracellular targets and display high protease resistance. Compared with small molecules, stapled peptides have larger surface areas, and therefore can selectively disrupt protein-protein interactions and have been successfully employed to modulate NOTCH and p53 signaling2.In the canonical Wnt/β-catenin signaling pathway, β-catenin stability is regulated by the destruction complex containing Axin, adenomatous polyposis coli (APC) and glycogen synthase kinase 3β (GSK3β). In the complex, Axin directly interacts with and targets β-catenin for GSK3β-mediated phosphorylation and subsequent ubiquitination and degradation3. To design cell-permeable stapled peptides that can activate Wnt/β-catenin signaling with good selectivity, we aim to disrupt the Axin-β-catenin interaction. The crystal structure of Axin reveals that its β-catenin-binding domain (Axin (469-482)) forms a continuous α-helix that fits into a shallow groove of β-catenin formed by the third helices of β-catenin armadillo repeats 3 and 44 (Figure 1A), suggesting that the Axin-β-catenin interaction is suitable for targeting by hydrocarbon-stapled α-helical peptide mimetics. We then synthesized the stapled peptides by incorporation of non-natural amino acids at neighboring positions along one face of the α-helix, followed by ring-closing olefin metathesis (Figure 1B). Two stapled α-helical peptides targeting the Axin-β-catenin complex (SAHPA), SAHPA1 and SAHPA2, were generated (Figure 1C). Circular dichroism analyses revealed that while Axin (469-482) displays 28% α-helical content, SAHPA1 and SAHPA2 have 34% and 57% α-helicity, respectively (Supplementary information, Figure S1).The binding of SAHPAs to β-catenin was investigated using an in vitro pull-down assay. Biotin-labeled SAHPAs were immobilized on streptavidin beads to pull down the purified glutathione-S-transferase (GST)-tagged fragment of β-catenin, GST-β-catenin (133-665). Both SAHPAs interacted directly with β-catenin, but SAHPA1 bound much stronger than SAHPA2 (Figure 1D). Quantification with isothermal titration calorimetry revealed that SAHPA1 bound to the purified β-catenin (133-665) with a dissociation constant of ~30.1 μM (Supplementary information, Figure S2).We then assessed cell permeability of the peptides by conjugating fluorescein isothiocyanate (FITC) to SAHPA1. Our data showed that FITC-SAHPA1 efficiently entered into the cytoplasm in Hela cells (Supplementary information, Figure S3). Subsequently we examined the ability of SAHPAs to interfere with the endogenous interaction between Axin and β-catenin in HEK293T cells. After immunoprecipitation of Axin2, the presence of β-catenin was detected by immunobloting. Axin-β-catenin association was greatly diminished by SAHPA1 only in the presence of Wnt3a (Figure 1E). SAHPA2 also had some effects, but its efficiency was much lower. Consistently, SAHPA1 greatly enhanced the effect of Wnt3a on stabilizing the active form of β-catenin (Figure 1F). Together, these experiments demonstrate that SAHPA1 binds to β-catenin and efficiently disrupts the endogenous Axin-β-catenin complex in the presence of Wnt3a.To explore the effects of SAHPAs on Wnt/β-catenin-dependent transcriptional activity, we performed the Topflash reporter assay in which the firefly luciferase is transcriptionally activated by β-catenin5. Consistent with the data above, SAHPA1 treatment greatly enhanced the reporter expression in a dose-dependent manner in the presence of Wnt3a (Figure 1G). SAHPA2 also promoted Wnt3a-induced reporter expression albeit less effectively than SAHPA1. Consistently, quantitative RT-PCR revealed that SAHPA1 increased the Wnt3a-stimulated expression of c-myc, axin2 and cyclin D1, the classic Wnt/β-catenin target genes6 (Figure 1H). In contrast, the unstapled fragment of Axin (469-482) had no enhancing effect. As controls, SAHPA1 did not affect other tested signaling pathway reporters (c-Jun-responsive AP-1 reporter, TGF-β/Smad-responsive CAGA reporter, BMP4-responsive BRE reporter, Notch/RBP-Jκ-responsive pGa981-6 reporter, TNFα-responsive NF-κB reporter and PKA-responsive CRE reporter) (Supplementary information, Figure S4), indicating the specific effect of SAHPA1 on Wnt/β-catenin signaling. These data together suggest that SAHPA1 efficiently activates Wnt/β-catenin signaling in a ligand-dependent manner.Wnt/β-catenin signaling has been shown to play important roles in the maintenance of self-renewal and prevention of differentiation of mouse embryonic stem cells (mESCs)7,8. We further investigated the effects of SAHPA1 on fate determination of R1 mESCs. R1 cells can be maintained in N2B27 medium containing LIF and BMP4, and withdrawal of these growth factors drives mESCs to neural differentiation9. Although Wnt3a alone could slightly reverse the neural differentiation process, Wnt3a plus SAHPA1 efficiently promoted self-renewal and inhibited differentiation of mESCs, as shown by colony morphology and alkaline phosphatase (AP) staining (Figure 1I). This observation was further supported by examining the expression of the pluripotency markers – Pou5f1 (Oct4), Nanog, and Rex1, and the neural markers – Sox1 and nestin10. Upon the withdrawal of LIF and BMP4, the expression of these pluripotency markers was significantly decreased and the expression of the neural markers was significantly increased (Figure 1J). SAHPA1 greatly enhanced the activities of Wnt3 to activate the pluripotency markers and to repress the neural markers. These results indicate that super activation of Wnt/β-catenin signaling by SAHPA1 plus Wnt3 could replace LIF and BMP4 to maintain mESC self-renewal.By far, most of the Wnt/β-catenin signaling agonists function through inhibition of GSK3β7,11. However, as GSK3β also plays important roles in metabolism and in the regulations of numerous signaling pathways12, such agonists could produce unwanted side effects. Therefore, Wnt/β-catenin agonists with high specificity would be appreciated for potential medical applications. Indeed, Gwak et al.13 recently reported the small molecule SKL2001, which can activate Wnt/β-catenin signaling even without Wnt treatment by disrupting the Axin-β-catenin interaction. Still, the specificity of SKL2001 waits to be tested. Due to the high binding specificity of stapled peptides, our approach can achieve high specificity as shown for SAHPA1 to activate the Wnt/β-catenin pathway without affecting other tested pathways.The potential carcinogenic effect of universal up-regulation of Wnt/β-catenin signailing greatly limits the clinical application of Wnt/β-catenin agonists. Our results reveal that the activation of Wnt/β-catenin signaling by SAHPA1 is ligand-dependent. In the absence of Wnt3a, SAHPA1 had no obvious effect. However, even with low levels of Wnt3a, SAHPA1 greatly amplified Wnt/β-catenin signaling. The ligand-dependent feature of SAHPA1 enables the activation of Wnt/β-catenin signaling in the tissues where Wnt is present, while not in the tissues without Wnt ligands, achieving another level of specificity.During the submission of our paper, Grossmann et al.14 reported that a hydrocarbon-stapled peptide (fStAx-35R) inhibited Wnt/β-catenin signaling by interfering with β-catenin-TCF interaction. To better discuss the difference between these two peptides, we compared their effects on Wnt/β-catenin signaling in HEK293T and colon cancer SW480 cells. fStAx-35R inhibited Wnt/β-catenin signaling in both cell lines. However, SAHPA1 enhanced Wnt/β-catenin signaling in HEK293T, but had no effect in SW480 cells, in which β-catenin is hyper-activated due to APC mutation and Wnt ligand has little effect (data not shown). Furthermore, SAHPA1 is exclusively localized in the cytoplasm of Hela cells (Supplementary information, Figure S3) and SW480 cells (data not shown), suggesting that the opposite effects of these two peptides could be due to their distinct subcellular localization (SAHPA1 in the cytoplasm and fStAx-35R in the nucleus14).In summary, we reported here a stapled α-helical peptide that targets the Axin-β-catenin interaction and activates Wnt/β-catenin signaling with a high selectivity. We have also demonstrated that this stapled peptide together with Wnt ligand could maintain the self-renewal of mouse embryonic stem cells in the absence of LIF and BMP. Our study provides a new strategy to modulate the Wnt/β-catenin pathway, and the highly specific agonist has potential applications in biomedical research and the treatment of Wnt/β-catenin signaling-defective diseases.We thank Dr Sheng-Cai Lin (School of Life Sciences, Xiamen University, China) for Axin2 antibody, Jianping Zhang and Yi Ding (School of Life Sciences, Tsinghua University, China) for technical assistance. This work was supported by grants from the 973 Program (2011CB943803 and 2011CB965300) and the National Natural Science Foundation of China (30930050, 91013007 and 30921004).(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Co-reporter:Hong-Kui Cui, Jie Qing, Ye Guo, Yu-Jia Wang, Li-Jia Cui, Tian-Hua He, Linqi Zhang, Lei Liu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 12) pp:3547-3554
Publication Date(Web):15 June 2013
DOI:10.1016/j.bmc.2013.02.011
The strategy of peptide stapling was used to develop new molecules to inhibit the hepatitis C virus infection via disrupting the binding of HCV envelope glycoprotein E2 with human cell surface protein CD81. The peptide sequence was designed based on the large extra-cellular loop of CD81 with known importance in the HCV E2 binding interaction. Our results showed that the stapled peptides exhibited significantly higher α-helicity and proteolytic stability as compared to their linear peptide counterpart. The optimal compound was found to have an EC50 value of ca. 17–39 μM against different HCV subtypes and represented a new HCV membrane fusion inhibitor.
Co-reporter:Qiao-Qiao He, Ge-Min Fang, Lei Liu
Chinese Chemical Letters 2013 Volume 24(Issue 4) pp:265-269
Publication Date(Web):April 2013
DOI:10.1016/j.cclet.2013.03.013
Protein chemical synthesis usually relies on the use of native chemical ligation that couples peptide thioester with a Cys-peptide. A limitation of this method is the difficulty of finding an appropriate Cys ligation site in many synthetic targets. To overcome this problem, the ligation–desulfurization approach has been developed. This approach involves the use of a thiol-containing amino acid as the ligation partner. After the sequence assembly is completed, the thiol group is removed through a desulfurization reaction to generate the standard amino acids. Currently this strategy has been applied to the ligations at a number of amino acids including Ala, Phe, Val, Lys, Thr, Leu, Pro and Gln. The present article reviews the design and synthesis of these thiol-containing amino acids for native chemical ligation at non-Cys sites.Recent advances in design and synthesis of thiol-containing amino acids in ligation–desulfurization strategy for native chemical ligation at non-Cys sites.
Co-reporter:Yi-Chao Huang;Dr. Yi-Ming Li;Dr. Yang Chen;Man Pan;Yi-Tong Li;Dr. Li Yu; Qing-Xiang Guo;Dr. Lei Liu
Angewandte Chemie 2013 Volume 125( Issue 18) pp:4958-4962
Publication Date(Web):
DOI:10.1002/ange.201209523
Co-reporter:Jun Yi;Xi Lu;Yan-Yan Sun;Bin Xiao; Lei Liu
Angewandte Chemie 2013 Volume 125( Issue 47) pp:12635-12639
Publication Date(Web):
DOI:10.1002/ange.201307069
Co-reporter:Jun Yi;Xi Lu;Yan-Yan Sun;Bin Xiao; Lei Liu
Angewandte Chemie International Edition 2013 Volume 52( Issue 47) pp:12409-12413
Publication Date(Web):
DOI:10.1002/anie.201307069
Co-reporter:Yi-Chao Huang;Dr. Yi-Ming Li;Dr. Yang Chen;Man Pan;Yi-Tong Li;Dr. Li Yu; Qing-Xiang Guo;Dr. Lei Liu
Angewandte Chemie International Edition 2013 Volume 52( Issue 18) pp:4858-4862
Publication Date(Web):
DOI:10.1002/anie.201209523
Co-reporter:Chu-Ting Yang ; Zhen-Qi Zhang ; Jun Liang ; Jing-Hui Liu ; Xiao-Yu Lu ; Huan-Huan Chen
Journal of the American Chemical Society 2012 Volume 134(Issue 27) pp:11124-11127
Publication Date(Web):June 26, 2012
DOI:10.1021/ja304848n
Practical catalytic cross-coupling of secondary alkyl electrophiles with secondary alkyl nucleophiles under Cu catalysis has been realized. The use of TMEDA and LiOMe is critical for the success of the reaction. This cross-coupling reaction occurs via an SN2 mechanism with inversion of configuration and therefore provides a general approach for the stereocontrolled formation of C–C bonds between two tertiary carbons from chiral secondary alcohols.
Co-reporter:Yiming Li, Maiyun Yang, Yichao Huang, Xiaoda Song, Lei Liu and Peng R. Chen
Chemical Science 2012 vol. 3(Issue 9) pp:2766-2770
Publication Date(Web):19 Jun 2012
DOI:10.1039/C2SC20433A
A series of alkene-bearing pyrrolysine analogues were synthesized and subsequently incorporated into proteins at two sites by a mutant PylRS–tRNA pair with excellent efficiency. This strategy allowed the site-specific labeling of proteins carrying single or double genetically encoded alkene handles via bioorthogonal thiol–ene ligation reactions.
Co-reporter:Jun Yi;Jin-Hui Liu;Jun Liang;Jian-Jun Dai;Chu-Ting Yang;Yao Fu
Advanced Synthesis & Catalysis 2012 Volume 354( Issue 9) pp:1685-1691
Publication Date(Web):
DOI:10.1002/adsc.201200136
Abstract
Palladium- and nickel-catalyzed cross-coupling recations of unactivated alkyl bromides with diboron reagents have been developed as practical methods for the synthesis of primary and secondary alkylboronic esters. These reactions extend the concept and utility of Pd- and Ni-catalyzed cross-coupling of aliphatic electrophiles. They also show different substrate selectivity and ligand dependence as compared to the recently reported Cu-catalyzed borylation reaction.
Co-reporter:Yi-Ming Li, Mai-Yun Yang, Yi-Chao Huang, Yi-Tong Li, Peng R. Chen, and Lei Liu
ACS Chemical Biology 2012 Volume 7(Issue 6) pp:1015
Publication Date(Web):March 16, 2012
DOI:10.1021/cb300020s
Expressed protein ligation bridges the gap between synthetic peptides and recombinant proteins and thereby significantly increases the size and complexity of chemically synthesized proteins. Although the intein-based expressed protein ligation method has been extensively used in this regard, the development of new expressed protein ligation methods may improve the flexibility and power of protein semisynthesis. In this study a new alternative version of expressed protein ligation is developed by combining the recently developed technologies of hydrazide-based peptide ligation and genetic code expansion. Compared to the previous intein-based expressed protein ligation method, the new method does not require the use of protein splicing technology and generates recombinant protein α-hydrazides as ligation intermediates that are more chemically stable than protein α-thioesters. Furthermore, the use of an evolved mutant pyrrolysyl-tRNA synthetase (PylRS), ACPK-RS, from M. barkeri shows an improved performance for the expression of recombinant protein backbone oxoesters. By using HdeA as a model protein we demonstrate that the hydrazide-based method can be used to synthesize proteins with correctly folded structures and full biological activity. Because the PylRS-tRNACUAPyl system is compatible with both prokaryotic and eukaryotic cells, the strategy presented here may be readily expanded to manipulate proteins produced in mammalian cells. The new hydrazide-based method may also supplement the intein-based expressed protein ligation method by allowing for a more flexible selection of ligation site.
Co-reporter:Chunlin Lv, Jin Zhang, Jian Hao, Lei Liu and Yongge Wei
Dalton Transactions 2012 vol. 41(Issue 33) pp:10065-10070
Publication Date(Web):18 May 2012
DOI:10.1039/C2DT30471F
Bi-alkylimido functionalization of hexamolybdates has been developed and several novel di-substituted alkylimido hexamolybdates with n-butyl, cyclohexyl and tert-butylamines as the imido-releasing reagents have been synthesized in reasonable yields. Their structures have been characterized by elemental analysis, IR, UV-Vis, and ESI mass spectrometry. Moreover, the structures of [Mo6O17(NCy)2]2− and [Mo6O17(NCMe3)2]2 have been determined by single-crystal X-ray diffraction techniques. It is notable that the compound (Bu4N)2[Mo6O17(NCy)2] crystallizes in polar space group Fdd2 with potential ferroelectricity. In addition, theoretical investigation of the reactivity and regioselectivity of bi-alkylimido functionalization has also been conducted. The calculation results show that more energy is required for the bi-functionalization process than for the mono-substitution process, and that the cis-bi-functionalization is the kinetically favored process while trans-[Mo6O17(NR)2]2− is the thermodynamically favored product.
Co-reporter:Chen Wang
Chinese Journal of Chemistry 2012 Volume 30( Issue 9) pp:1974-1979
Publication Date(Web):
DOI:10.1002/cjoc.201200337
Abstract
Imidazole-promoted ligation of peptide phenyl esters was recently found to be a complementary method for protein chemical synthesis. Theoretical calculations have been carried out to understand the detailed mechanism of this particular ligation process. It is found that both the reaction of the phenyl ester with imidazole and the reaction of the acyl imidazole intermediate with cysteine proceed through an addition-elimination mechanism. The cleavage of the CO bond in the reaction between the phenyl ester and imidazole is the rate-limiting step of the overall ligation process. Interestingly, although the imidazole-promoted phenyl ester ligation has a higher free energy barrier than the conventional thiophenol-promoted native chemical ligation for a sterically less hindered C-terminal amino acid (e.g. gylcine), for a sterically hindered C-terminal amino acid (e.g. proline) the imidazole-promoted phenyl ester ligation is calculated to be more favorable than the conventional thiophenol-promoted native chemical ligation.
Co-reporter:Ji-Shen Zheng;Shan Tang;Ye Guo;Hao-Nan Chang; Dr. Lei Liu
ChemBioChem 2012 Volume 13( Issue 4) pp:542-546
Publication Date(Web):
DOI:10.1002/cbic.201100580
Co-reporter:Dr. Ge-Min Fang;Jia-Xing Wang;Dr. Lei Liu
Angewandte Chemie 2012 Volume 124( Issue 41) pp:10493-10496
Publication Date(Web):
DOI:10.1002/ange.201203843
Co-reporter:YuanYe Jiang;Yao Fu
Science China Chemistry 2012 Volume 55( Issue 10) pp:2057-2062
Publication Date(Web):2012 October
DOI:10.1007/s11426-012-4672-0
Recently we reported Pd-catalyzed decarboxylative cross-coupling of cyanoacetate salts with aryl halides and triflates. This reaction shows good functional group tolerance and is useful for the synthesis of α-aryl nitriles. To elucidate the mechanism for this reaction, we now carry out a density functional theory study on the cross-coupling of potassium cyanoacetate with bromobenzene. Our results show that the decarboxylation transition state involving the interaction of Pd with the α-carbon atom has a very high energy barrier of +34.5 kcal/mol and therefore, must be excluded. Decarboxylation of free ion (or tight-ion-pair) also causes a high energy increase and should be ruled out. Thus the most favored decarboxylation mechanism corresponds to a transition state in which Pd interacts with the cyano nitrogen. The energy profile of the whole catalytic cycle shows that decarboxylation is the rate-determining step. The total energy barrier is +27.5 kcal/mol, which is comprised of two parts, i.e. the energy barrier for decarboxylation and the energy cost for transmetallation.
Co-reporter:Dr. Ge-Min Fang;Jia-Xing Wang;Dr. Lei Liu
Angewandte Chemie International Edition 2012 Volume 51( Issue 41) pp:10347-10350
Publication Date(Web):
DOI:10.1002/anie.201203843
Co-reporter:Chu-Ting Yang;Zhen-Qi Zhang;Hazmi Tajuddin;Chen-Cheng Wu;Jun Liang;Jing-Hui Liu;Yao Fu;Maria Czyzewska;Dr. Patrick G. Steel;Dr. Todd B. Marder;Dr. Lei Liu
Angewandte Chemie International Edition 2012 Volume 51( Issue 2) pp:528-532
Publication Date(Web):
DOI:10.1002/anie.201106299
Co-reporter:Ji-Shen Zheng;Hao-Nan Chang;Jing Shi
Science China Chemistry 2012 Volume 55( Issue 1) pp:64-69
Publication Date(Web):2012 January
DOI:10.1007/s11426-011-4434-4
Cyclotides constitute a fascinating family of circular proteins containing ca. 30 amino acid residues. They have a unique cyclic cysteine knot topology and exhibit remarkable thermal, chemical and enzymatic stabilities. These characteristics enable them to have a range of biological activities and promising pharmaceutical and agricultural applications. Here, we present a practical strategy for the chemical synthesis of cyclotides through the intramolecular ligation of fully unprotected peptide O-esters. This strategy involves the mild Fmoc solid-phase peptide synthesis of the peptide O-ester backbone, the head-to-tail cyclization of the cyclotide backbone by native chemical ligation, and the oxidative refolding to yield the natural knot protein. The simplicity and high efficiency of the strategy can be employed in the synthesis of artificial cyclotides for pharmaceutical applications.
Co-reporter:Bin Xiao ; Tian-Jun Gong ; Jun Xu ; Zhao-Jing Liu
Journal of the American Chemical Society 2011 Volume 133(Issue 5) pp:1466-1474
Publication Date(Web):January 10, 2011
DOI:10.1021/ja108450m
Pd-catalyzed directed ortho C−H amidation of aromatic ketones with both sulfonamides and amides has been accomplished. The use of an electron-deficient Pd complex, Pd(OTf)2, is crucial for the success of this transformation. Some key intermediates of the reaction, that is, the cyclopalladation complexes of ketones, have been characterized by X-ray crystallography. Experimental analysis of these palladacycles and also the experimental results with N-methyl sulfonamides indicate that the new reaction does not seem to proceed through a nitrene intermediate. The utility of the newly developed reaction was demonstrated for the synthesis of useful organic intermediates such as 2- and 3-alkyl indoles and 2-aminophenyl ketones.
Co-reporter:Bin Xiao ; Tian-Jun Gong ; Zhao-Jing Liu ; Jing-Hui Liu ; Dong-Fen Luo ; Jun Xu
Journal of the American Chemical Society 2011 Volume 133(Issue 24) pp:9250-9253
Publication Date(Web):May 24, 2011
DOI:10.1021/ja203335u
A practical, Pd(0)/Pd(II)-catalyzed reaction was developed for phenol-directed C–H activation/C–O cyclization using air as an oxidant. The turnover-limiting step of the process was found to be C–O reductive elimination instead of C–H activation. This reaction can tolerate a variety of functional groups and is complementary to the previous methods for the synthesis of substituted dibenzofurans.
Co-reporter:Ji-Shen Zheng ; Hao-Nan Chang ; Feng-Liang Wang
Journal of the American Chemical Society 2011 Volume 133(Issue 29) pp:11080-11083
Publication Date(Web):June 29, 2011
DOI:10.1021/ja204088a
An operationally simple method for the synthesis of peptide thioesters is developed using standard Fmoc solid-phase peptide synthesis procedures. The method relies on the use of a premade enamide-containing amino acid which, in the final TFA cleavage step, renders the desired thioester functionality through an irreversible intramolecular N-to-S acyl transfer.
Co-reporter:Jun Xu, Yao Fu, Dong-Fen Luo, Yuan-Ye Jiang, Bin Xiao, Zhao-Jing Liu, Tian-Jun Gong, and Lei Liu
Journal of the American Chemical Society 2011 Volume 133(Issue 39) pp:15300-15303
Publication Date(Web):September 13, 2011
DOI:10.1021/ja206330m
An unprecedented type of reaction for Cu-catalyzed trifluoromethylation of terminal alkenes is reported. This reaction represents a rare instance of catalytic trifluoromethylation through C(sp3)–H activation. It also provides a mechanistically unique example of Cu-catalyzed allylic C–H activation/functionalization. Both experimental and theoretical analyses indicate that the trifluoromethylation may occur via a Heck-like four-membered-ring transition state.
Co-reporter:Jun Xu, Dong-Fen Luo, Bin Xiao, Zhao-Jing Liu, Tian-Jun Gong, Yao Fu and Lei Liu
Chemical Communications 2011 vol. 47(Issue 14) pp:4300-4302
Publication Date(Web):07 Mar 2011
DOI:10.1039/C1CC10359H
A copper-catalyzed process for trifluoromethylation of aryl, heteroaryl, and vinyl boronic acids has been developed. The reaction is conducted under mild conditions and shows tolerance to moisture and a variety of functional groups.
Co-reporter:Jian-Jun Dai, Jing-Hui Liu, Dong-Fen Luo and Lei Liu
Chemical Communications 2011 vol. 47(Issue 2) pp:677-679
Publication Date(Web):24 Nov 2010
DOI:10.1039/C0CC04104A
Pd-catalysed decarboxylative Suzuki reactions and orthogonal Cu-based O-arylation reactions of aromatic carboxylic acids are reported. The new reactions may provide alternative routes for the synthesis of some biaryls and aromatic carboxylic esters.
Co-reporter:Fei Shen;Yi-Chao Huang;Shan Tang;Yong-Xiang Chen
Israel Journal of Chemistry 2011 Volume 51( Issue 8-9) pp:940-952
Publication Date(Web):
DOI:10.1002/ijch.201100076
Abstract
The development of efficient chemical methods for total synthesis or semisynthesis of integral membrane proteins is an important challenge at the interface between chemistry and biology. This review outlines the recent advances in the synthesis of integral membrane proteins, with particular focus on the methods for difficult peptide synthesis, purification, and enhancement of peptide solubility under the ligation conditions. The applications of these methods to the synthesis of integral membrane proteins with one or multiple transmembrane domains are also described.
Co-reporter:Chu-Ting Yang;Zhen-Qi Zhang;Yu-Chen Liu ;Dr. Lei Liu
Angewandte Chemie 2011 Volume 123( Issue 17) pp:3990-3993
Publication Date(Web):
DOI:10.1002/ange.201008007
Co-reporter:Rui Shang;Dong-Sheng Ji;Ling Chu;Dr. Yao Fu;Dr. Lei Liu
Angewandte Chemie 2011 Volume 123( Issue 19) pp:4562-4566
Publication Date(Web):
DOI:10.1002/ange.201006763
Co-reporter:Ge-Min Fang;Yi-Ming Li;Fei Shen;Yi-Chao Huang;Jia-Bin Li;Yun Lin;Hong-Kui Cui ;Dr. Lei Liu
Angewandte Chemie 2011 Volume 123( Issue 33) pp:7787-7791
Publication Date(Web):
DOI:10.1002/ange.201100996
Co-reporter:Chu-Ting Yang;Zhen-Qi Zhang;Yu-Chen Liu ;Dr. Lei Liu
Angewandte Chemie International Edition 2011 Volume 50( Issue 17) pp:3904-3907
Publication Date(Web):
DOI:10.1002/anie.201008007
Co-reporter:Fei Shen;Shan Tang
Science China Chemistry 2011 Volume 54( Issue 1) pp:110-116
Publication Date(Web):2011 January
DOI:10.1007/s11426-010-4188-4
The study on membrane proteins is an important challenge mainly because of their very poor solubility in various solvents. The traditional recombinant expression strategy and the native chemical ligation method both have difficulty in generating sufficient amounts of desired proteins with high efficiency. Previous studies have shown that multiply fluorinated alcohols exhibit good ability to dissolve difficult peptide sequences, especially hexafluoro-2-propanol (HFIP). In the present study we systematically studied the capability of solvents containing different percentage of HFIP in dissolving transmembrane peptides. Through both HPLC and UV analyses we concluded that 60% HFIP/8 M urea constituted a good solvent system. In this solvent system we also optimized conditions to perform native chemical ligation (NCL). Under the optimized conditions we successfully achieved NCL’s for both dipeptide formation and the synthesis of a model protein (Trifolitoxin). These results suggested that HFIP was a potential cosolvent that could be used in the ligation of poorly soluble peptides for the generation of membrane proteins.
Co-reporter:Rui Shang
Science China Chemistry 2011 Volume 54( Issue 11) pp:1670-1687
Publication Date(Web):2011 November
DOI:10.1007/s11426-011-4381-0
Transition metal-catalyzed decarboxylative cross-coupling reactions have recently emerged as a new and important category of organic transformations that find versatile applications in the construction of carbon-carbon and carbon-heteroatom bonds. The use of relatively cheap and stable carboxylic acids to replace organometallic reagents enables the decarboxylative cross-coupling reactions to proceed with good selectivities and functional group tolerance. In the present review we summarize the various types of decarboxylative cross-coupling reactions catalyzed by different transition metal complexes. The scope and applications of these reactions are described. The challenges and opportunities in the field are discussed.
Co-reporter:Rui Shang;Dong-Sheng Ji;Ling Chu;Dr. Yao Fu;Dr. Lei Liu
Angewandte Chemie International Edition 2011 Volume 50( Issue 19) pp:4470-4474
Publication Date(Web):
DOI:10.1002/anie.201006763
Co-reporter:Ge-Min Fang;Yi-Ming Li;Fei Shen;Yi-Chao Huang;Jia-Bin Li;Yun Lin;Hong-Kui Cui ;Dr. Lei Liu
Angewandte Chemie International Edition 2011 Volume 50( Issue 33) pp:7645-7649
Publication Date(Web):
DOI:10.1002/anie.201100996
Co-reporter:Rui Shang ; Zhi-Wei Yang ; Yan Wang ; Song-Lin Zhang
Journal of the American Chemical Society 2010 Volume 132(Issue 41) pp:14391-14393
Publication Date(Web):September 28, 2010
DOI:10.1021/ja107103b
Pd-catalyzed decarboxylative cross-couplings of 2-(2-azaaryl)acetates with aryl halides and triflates have been discovered. This reaction is potentially useful for the synthesis of some functionalized pyridines, quinolines, pyrazines, benzoxazoles, and benzothiazoles. Theoretical analysis shows that the nitrogen atom at the 2-position of the heteroaromatics directly coordinates to Pd(II) in the decarboxylation transition state.
Co-reporter:Rui Shang, Qing Xu, Yuan-Ye Jiang, Yan Wang and Lei Liu
Organic Letters 2010 Volume 12(Issue 5) pp:1000-1003
Publication Date(Web):January 29, 2010
DOI:10.1021/ol100008q
Pd-catalyzed decarboxylative cross coupling of potassium polyfluorobenzoates with aryl bromides, chlorides, and triflates is achieved by using diglyme as the solvent. The reaction is useful for synthesis of polyfluorobiaryls from readily accessible and nonvolatile polyfluorobenzoate salts. Unlike the Cu-catalyzed decarboxylation cross coupling where oxidative addition is the rate-limiting step, in the Pd-catalyzed version decarboxylation is the rate-limiting step.
Co-reporter:Juan Li, Hong-Kui Cui, Lei Liu
Tetrahedron Letters 2010 Volume 51(Issue 13) pp:1793-1796
Publication Date(Web):31 March 2010
DOI:10.1016/j.tetlet.2010.01.108
New thiol-containing auxiliaries were developed for peptide ligation. They were placed at the amidyl N-atom in the second amino acid residue of a peptide fragment. With the new auxiliaries, peptide ligation could be conducted at non-Cys and non-Gly sites. Compared to other recently developed auxiliaries, an important feature of the present design was that the new auxiliaries were generally applicable and readily removable.
Co-reporter:Yi-Ming Li, Jing Shi, Rong Cai, Xiao-Yun Chen, Qing-Xiang Guo, Lei Liu
Tetrahedron Letters 2010 Volume 51(Issue 12) pp:1609-1612
Publication Date(Web):24 March 2010
DOI:10.1016/j.tetlet.2010.01.071
A series of quinoline-based photo-removable protecting (caging) groups were synthesized for the development of new chemical tools to photo-regulate bioactive molecules in living cells and tissues with improved properties. Compared with the recently developed 8-bromo-7-hydroxyquinolinyl (BHQ) chromophore, change of the bromine substituent to a pyridine group led to a new photo-labile group (3′-PyHQ) with an increased water solubility, a lower self-fluorescence, and a higher photolysis efficiency. It was proposed that the replacement of a halogen group by a pyridine-like heterocycle may provide a general strategy to improve the existing photo-caging groups.
Co-reporter:Zhe Li Dr.;Yao Fu ;Song-Lin Zhang;Qing-Xiang Guo
Chemistry – An Asian Journal 2010 Volume 5( Issue 6) pp:1475-1486
Publication Date(Web):
DOI:10.1002/asia.200900744
Abstract
The mechanism of a recently discovered intramolecular Heck-type coupling of oximes with aryl halides (Angew. Chem. Int. Ed. 2007, 46, 6325) was systematically studied by using density functional methods enhanced with a polarized continuum solvation model. The overall catalytic cycle of the reaction was found to consist of four steps: oxidative addition, migratory insertion, β-H elimination, and catalyst regeneration, whereas an alternative base-promoted CH activation pathway was determined to be less favorable. Migratory insertion was found to be the rate determining step in the catalytic cycle. The apparent activation barrier of migratory insertion of the (E)-oxime was +20.5 kcal mol−1, whereas the barrier of (Z)-oxime was as high as +32.7 kcal mol−1. However, (Z)-oxime could isomerize to form the more active (E)-oxime with the assistance of K2CO3, so that both the (E)- and (Z)-oxime substrates could be transformed to the desired product. Our calculations also indicated that the Z product was predominant in the equilibrium of the isomerization of the imine double bond, which constituted the reason for the good Z-selectivity observed for the reaction. Furthermore, we examined the difference between the intermolecular Heck-type reactions of imines and of olefins. It was found that in the intermolecular Heck-type coupling of imines, the apparent activation barrier of migratory insertion was as high as +35 kcal mol−1, which should be the main obstacle of the reaction. The analysis also revealed the main problem for the intermolecular Heck-type reactions of imines, which was that the breaking of a CN π bond was much more difficult than the breaking of a CC π bond. After systematic examination of a series of substituted imines, (Z)-N-amino imine and N-acetyl imine were found to have relatively low barriers of migratory insertion, so that they might be possible substrates for intermolecular Heck-type coupling.
Co-reporter:Ji-Shen Zheng;Hong-Kui Cui;Ge-Min Fang;Wei-Xian Xi
ChemBioChem 2010 Volume 11( Issue 4) pp:511-515
Publication Date(Web):
DOI:10.1002/cbic.200900789
Co-reporter:Ge-Min Fang;Hong-Kui Cui;Ji-Shen Zheng Dr.
ChemBioChem 2010 Volume 11( Issue 8) pp:1061-1065
Publication Date(Web):
DOI:10.1002/cbic.201000165
Co-reporter:Chen Wang;Yao Fu ;Qing-Xiang Guo
Chemistry - A European Journal 2010 Volume 16( Issue 8) pp:2586-2598
Publication Date(Web):
DOI:10.1002/chem.200902484
Abstract
Quantitative nucleophilicity scales are fundamental to organic chemistry and are usually constructed on the basis of Mayr’s equation [log k=s(N+E)] by using benzhydrylium ions as reference electrophiles. Here an ab initio protocol was developed for the first time to predict the nucleophilicity parameters N of various π nucleophiles in CH2Cl2 through transition-state calculations. The optimized theoretical model (BH&HLYP/6-311++G(3df,2p)//B3LYP/6-311+G(d,p)/PCM/UAHF) could predict the N values of structurally unrelated π nucleophiles within a precision of ca. 1.14 units and therefore may find applications for the prediction of nucleophilicity of compounds that are not readily amenable to experimental characterization. The success in predicting N parameters from first principles also allowed us to analyze in depth the electrostatic, steric, and solvation energies involved in electrophile–nucleophile reactions. We found that solvation does not play an important role in the validity of Mayr’s equation. On the other hand, the correlations of the E, N, and log k values with the energies of the frontier molecular orbitals indicated that electrostatic/charge-transfer interactions play vital roles in Mayr’s equation. Surprising correlations observed between the electrophile–nucleophile CC distances in the transition state, the activation energy barriers, and the E and N parameters indicate the importance of steric interactions in Mayr’s equation. A method is then proposed to separate the attraction and repulsion energies in the nucleophile–electrophile interaction. It was found that the attraction energy correlated with N+E, whereas the repulsion energy correlated to the s parameter.
Co-reporter:Song-Lin Zhang ; Yao Fu ; Rui Shang ; Qing-Xiang Guo
Journal of the American Chemical Society 2009 Volume 132(Issue 2) pp:638-646
Publication Date(Web):December 28, 2009
DOI:10.1021/ja907448t
Transition-metal-catalyzed decarboxylative coupling presents a new and important direction in synthetic chemistry. Mechanistic studies on decarboxylative coupling not only improve the understanding of the newly discovered transformations, but also may have valuable implications for the development of more effective catalyst systems. In this work, a comprehensive theoretical study was conducted on the mechanism of Myers’ Pd-catalyzed decarboxylative Heck reaction. The catalytic cycle was found to comprise four steps: decarboxylation, olefin insertion, β-hydride elimination, and catalyst regeneration. Decarboxylation was the rate-limiting step, and it proceeded through a dissociative pathway in which Pd(II) mediated the extrusion of CO2 from an aromatic carboxylic acid to form a Pd(II)−aryl intermediate. Further analysis was conducted on the factors that might control the efficiency of Myers’ decarboxylative Heck reaction. These factors included Pd salts, ligands, acid substrates, and metals. (1) Regarding Pd salts, PdCl2 and PdBr2 were worse catalysts than Pd(TFA)2, because the exchange of Cl or Br by a carboxylate from Pd was thermodynamically unfavorable. (2) Regarding ligands, DMSO provided the best compromise between carboxyl exchange and decarboxylation. Phosphines and N-heterocarbenes disfavored decarboxylation because of their electron richness, whereas pyridine ligands disfavored carboxyl exchange. (3) Regarding acid substrates, a good correlation was observed between the energy barrier of R−COOH decarboxylation and the R−H acidity. Substituted benzoic acids showed deviation from the correlation because of the involvement of π(substituent)−σ(Cipso−Pd) interaction. (4) Regarding metals, Ni and Pt were worse catalysts than Pd because of the less favorable carboxyl exchange and/or DMSO removal steps in Ni and Pt catalysis.
Co-reporter:Zhe Li ; Song-Lin Zhang ; Yao Fu ; Qing-Xiang Guo
Journal of the American Chemical Society 2009 Volume 131(Issue 25) pp:8815-8823
Publication Date(Web):June 8, 2009
DOI:10.1021/ja810157e
Ni-catalyzed selective C−O bond activation opens a door for the cross-coupling of aryl esters. The present study reports a thorough theoretical analysis of Ni-catalyzed cross-coupling between aryl esters and arylboronic acids, with an emphasis on explaining the cause for the surprising selectivity in C−O activation. The overall catalytic cycle is found to include three basic steps: oxidative addition, transmetalation, and reductive elimination. Oxidative addition of Ar−OAc to Ni(0) in the presence of PCy3 ligand proceeds through the monophosphine pathway (instead of the alternative two-phosphine pathway) with a relatively low barrier of +22.9 kcal/mol. Transmetalation proceeds via a base-assisted mechanism with a barrier of +31.2 kcal/mol. Reductive elimination is the most facile step in the whole catalytic cycle. Comparatively, oxidative addition of ArO−Ac to Ni(0) is a more facile process (barrier = +14.2 kcal/mol) than oxidative addition of Ar−OAc to Ni(0). However, the former process is associated with a fairly low reverse barrier, and its product does not transmetalate easily (barrier = +33.1 kcal/mol). By comparison, the latter process is an irreversible reaction, and its product transmetalates more readily. These results explain why only the cross-coupling products from the Ar−OAc activation (but not from the ArO−Ac activation) were observed in experiments.
Co-reporter:Rui Shang ; Yao Fu ; Jia-Bin Li ; Song-Lin Zhang ; Qing-Xiang Guo
Journal of the American Chemical Society 2009 Volume 131(Issue 16) pp:5738-5739
Publication Date(Web):April 1, 2009
DOI:10.1021/ja900984x
Pd-catalyzed decarboxylative cross-coupling of aryl iodides, bromides, and chlorides with potassium oxalate monoesters has been discovered. This reaction is potentially useful for laboratory-scale synthesis of aryl and alkenyl esters. Bulky, electron-rich bidentate phosphine ligands are preferred in the reaction, whereas Cu is not needed for decarboxylation. Theoretical calculations suggest a five-coordinate Pd(II) transition state for decarboxylation with an energy barrier of ∼30 kcal/mol.
Co-reporter:Xiao-Yun Chen, Jing Shi, Yi-Ming Li, Feng-Liang Wang, Xu Wu, Qing-Xiang Guo and Lei Liu
Organic Letters 2009 Volume 11(Issue 19) pp:4426-4429
Publication Date(Web):September 1, 2009
DOI:10.1021/ol901787w
A new fluorescent probe for monitoring Zn2+ was synthesized based on the structure of 7-hydroxyquinoline. Compared with 8-substituted quinolines, the new probe exhibited higher selectivity for Zn2+ over Cd2+. Its fluorescence enhancement (14-fold) and nanomolar range sensitivity (Kd = 0.117 nM) were favorable toward biological applications. Experiments also showed that a cell-permeable derivative of the new probe was potentially useful for two-photon imaging in living cells.
Co-reporter:Rui Shang;Yao Fu Dr.;Yan Wang;Qing Xu;Hai-Zhu Yu Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 49) pp:9350-9354
Publication Date(Web):
DOI:10.1002/anie.200904916
Co-reporter:Chu-Ting Yang;Yao Fu ;Yao-Bing Huang;Jun Yi;Qing-Xiang Guo
Angewandte Chemie International Edition 2009 Volume 48( Issue 40) pp:7398-7401
Publication Date(Web):
DOI:10.1002/anie.200903158
Co-reporter:Yang Zhou;Jing Zhao Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 39) pp:7126-7128
Publication Date(Web):
DOI:10.1002/anie.200902762
Co-reporter:Rui Shang;Yao Fu Dr.;Yan Wang;Qing Xu;Hai-Zhu Yu Dr.
Angewandte Chemie 2009 Volume 121( Issue 49) pp:9514-9518
Publication Date(Web):
DOI:10.1002/ange.200904916
Co-reporter:Zhe Li ; Yao Fu ; Qing-Xiang Guo
Organometallics 2008 Volume 27(Issue 16) pp:4043-4049
Publication Date(Web):July 22, 2008
DOI:10.1021/om701065f
The mechanism of oxidative addition of aryl halides to Pd(PR3)2 (R = Me, Et, iPr, tBu, Ph) was investigated by using density functional theory methods enhanced with a polarized continuum solvation model. Different reaction pathways were discussed on the basis of Gibbs free-energy profiles in a tetrahydrofuran solution. The calculations indicated that monophosphine PdPR3 was catalytically more active than bisphosphine Pd(PR3)2 for oxidative addition. However, among different PR3 ligands (R = Me, Et, iPr, tBu, Ph), the free-energy barriers for oxidative addition to PdPR3 did not change significantly (i.e., less than 2 kcal/mol). This gave rise to an important question: why was P(t-Bu)3 the only catalytically active ligand toward aryl chlorides among the above five ligands? It was proposed on the basis of the calculated data that the difference of the dissociation energies from PdL2 to PdL and L (L = ligand) between the various PR3 ligands dictated their dissimilar reactivity in oxidative addition.
Co-reporter:Yao Fu ; Zhe Li ; Shuang Liang ; Qing-Xiang Guo
Organometallics 2008 Volume 27(Issue 15) pp:3736-3742
Publication Date(Web):July 9, 2008
DOI:10.1021/om800067u
Carbon−oxygen bond-forming reductive elimination from transient Pd(IV) aryl/acetate complexes was recently implicated as the product release step in Pd(II)-catalyzed arene oxygenation reactions. The mechanistic details of C−O bond formation from these Pd(IV) intermediates remain elusive and, therefore, are subjected to a systematic theoretical investigation in the present study. Three proposed mechanisms are examined including (A) pre-equilibrium dissociation of a benzoate ligand followed by reductive elimination from the resulting five-coordinate Pd(IV); (B) direct reductive elimination from the six-coordinate Pd(IV); and (C) dissociation of a pyridyl arm of one cyclometalated ligand followed by internal coupling. Through density functional theory calculations it is suggested that mechanism B is favored over the other two mechanisms. This conclusion is supported by the success of the theoretical model based on mechanism B to reproduce the experimental activation free energy barriers. The same theoretical model also can reproduce the small effect of the solvent polarity and the negative Hammett reaction constant associated with the reductive elimination rates. All of these results suggest that mechanism A should not be involved in the reductive elimination. Furthermore, our calculations explain why the rate of reductive elimination from the Pd(IV) complex of bisphenylpyridine is significantly faster than that from the Pd(IV) complex of bisbenzo[h]quinoline.
Co-reporter:Xiu-Juan Qi, Zhe Li, Yao Fu, Qing-Xiang Guo and Lei Liu
Organometallics 2008 Volume 27(Issue 12) pp:2688-2698
Publication Date(Web):May 20, 2008
DOI:10.1021/om701135c
The homolytic Co−C bond dissociation enthalpy (BDE) is central to the understanding of organocobalt-mediated reactions in the areas of both bioinorganic chemistry and transition-metal catalysis. However, the determination of the Co−C BDEs still remains a difficult task using either an experimental or theoretical approach. Here we investigate how to use the density functional theory method to accurately calculate the Co−C BDEs by testing a number of functionals. It is found that the recently developed TPSS/LANL2DZ+p method can reproduce 28 experimental Co−C BDEs within a precision of ca. 2.2 kcal/mol. Equipped with this useful tool, we next examined the effects of the in-plane ligands on the Co−C BDEs in a systematic fashion for the first time. It is found that the in-plane ligands can vary the Co−C BDEs by ca. 10 kcal/mol. Across different in-plane ligands the Co−C BDEs are found to exhibit a strong, negative correlation with the spin densities at the cobalt atoms after the homolysis. This observation is not consistent with the conventional chemical intuition that delocalization of the spin of a free radical through the hyperconjugation interactions should stabilize the radical and, thereby, weaken the chemical bond that undergoes homolysis. We name this unexpected finding the anti-spin-delocalization effect. Further analyses of the molecular orbitals and atomic charges indicate that (1) the in-plane ligands can reduce the Co spin density through hyperconjugation with their empty antibonding π* orbitals and (2) the in-plane ligands can also stabilize the Co−C starting material through ionic interactions by attracting electrons from Co. Both the stabilization effects are determined by the electronegativity of the in-plane ligands. Thus the origin for the anti-spin-delocalization effect is proposed to be that the stabilization effect of the in-plane ligands is larger for the starting material than for the radical.
Co-reporter:Ji-Shen Zheng; Yao He; Chao Zuo; Xiao-Ying Cai; Shan Tang; Zhipeng A. Wang; Long-Hua Zhang; Chang-Lin Tian
Journal of the American Chemical Society () pp:
Publication Date(Web):March 4, 2016
DOI:10.1021/jacs.6b00515
Chemical protein synthesis can provide access to proteins with post-translational modifications or site-specific labelings. Although this technology is finding increasing applications in the studies of water-soluble globular proteins, chemical synthesis of membrane proteins remains elusive. In this report, a general and robust removable backbone modification (RBM) method is developed for the chemical synthesis of membrane proteins. This method uses an activated O-to-N acyl transfer auxiliary to install in the Fmoc solid-phase peptide synthesis process a RBM group with switchable reactivity toward trifluoroacetic acid. The method can be applied to versatile membrane proteins because the RBM group can be placed at any primary amino acid. With RBM, the membrane proteins and their segments behave almost as if they were water-soluble peptides and can be easily handled in the process of ligation, purification, and mass characterizations. After the full-length protein is assembled, the RBM group can be readily removed by trifluoroacetic acid. The efficiency and usefulness of the new method has been demonstrated by the successful synthesis of a two-transmembrane-domain protein (HCV p7 ion channel) with site-specific isotopic labeling and a four-transmembrane-domain protein (multidrug resistance transporter EmrE). This method enables practical synthesis of small- to medium-sized membrane proteins or membrane protein domains for biochemical and biophysical studies.
Co-reporter:Bin Xiao ; Yao Fu ; Jun Xu ; Tian-Jun Gong ; Jian-Jun Dai ; Jun Yi
Journal of the American Chemical Society () pp:
Publication Date(Web):December 18, 2009
DOI:10.1021/ja909818n
Although nitrogen-containing group-directed cyclopalladation reactions have been well-known, Pd(II) insertion into C−H bonds promoted by coordination of an oxygen-only group to the palladium remains rather rare. In the present study, the first cyclopalladation complex formed from a simple phenol ester was characterized by X-ray crystallography. A promising protocol for the ortho C−H activation/aryl−aryl coupling of phenol esters that was not sensitive to moisture or air was then established. The utility of the reaction was demonstrated for the synthesis of useful phenol derivatives.
Co-reporter:Jian-Jun Dai, Jing-Hui Liu, Dong-Fen Luo and Lei Liu
Chemical Communications 2011 - vol. 47(Issue 2) pp:NaN679-679
Publication Date(Web):2010/11/24
DOI:10.1039/C0CC04104A
Pd-catalysed decarboxylative Suzuki reactions and orthogonal Cu-based O-arylation reactions of aromatic carboxylic acids are reported. The new reactions may provide alternative routes for the synthesis of some biaryls and aromatic carboxylic esters.
Co-reporter:Chunlin Lv, Jin Zhang, Jian Hao, Lei Liu and Yongge Wei
Dalton Transactions 2012 - vol. 41(Issue 33) pp:NaN10070-10070
Publication Date(Web):2012/05/18
DOI:10.1039/C2DT30471F
Bi-alkylimido functionalization of hexamolybdates has been developed and several novel di-substituted alkylimido hexamolybdates with n-butyl, cyclohexyl and tert-butylamines as the imido-releasing reagents have been synthesized in reasonable yields. Their structures have been characterized by elemental analysis, IR, UV-Vis, and ESI mass spectrometry. Moreover, the structures of [Mo6O17(NCy)2]2− and [Mo6O17(NCMe3)2]2 have been determined by single-crystal X-ray diffraction techniques. It is notable that the compound (Bu4N)2[Mo6O17(NCy)2] crystallizes in polar space group Fdd2 with potential ferroelectricity. In addition, theoretical investigation of the reactivity and regioselectivity of bi-alkylimido functionalization has also been conducted. The calculation results show that more energy is required for the bi-functionalization process than for the mono-substitution process, and that the cis-bi-functionalization is the kinetically favored process while trans-[Mo6O17(NR)2]2− is the thermodynamically favored product.
Co-reporter:Jiabin Li, Yuanyuan Li, Qiaoqiao He, Yiming Li, Haitao Li and Lei Liu
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 29) pp:NaN5441-5441
Publication Date(Web):2014/06/17
DOI:10.1039/C4OB00715H
One of the rising demands in the field of protein chemical synthesis is the development of facile strategies that yield the protein in workable quantities and homogeneity, with fewer handling steps. Although the native chemical ligation of peptide hydrazides has recently been shown to be useful for the chemical synthesis of proteins carrying acid-sensitive modification groups, previous hydrazide-based protein synthesis studies have used sequential ligation strategies. Here, we report a practical method for a “one-pot” native chemical ligation of peptide hydrazides that would circumvent the need for the isolation of the intermediate products. This method employed a fast and selective arylboronate oxidation reaction mediated by H2O2, which draws attention to the potential applications of the thus far under-exploited boron-based functionalities in protein chemical synthesis. To demonstrate the practicality and efficiency of the new one-pot method, we report its application to a scalable total synthesis of modified histones (with five analogues of H3 and H4 as examples) on a multi-milligram scale, with good homogeneity.
Co-reporter:Yun-Kun Qi, Shan Tang, Yi-Chao Huang, Man Pan, Ji-Shen Zheng and Lei Liu
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 18) pp:NaN4198-4198
Publication Date(Web):2016/04/14
DOI:10.1039/C6OB00450D
A new thiol protecting group Hmboff/on is described, which has a switchable activity that may be useful in the chemical synthesis of proteins. When placed on the side chain of Cys, Cys(Hmboff) is stable to trifluoroacetic acid (TFA) in the process of solid-phase peptide synthesis. When Cys(Hmboff) is treated with neutral aqueous buffers, it is cleanly converted to acid-labile Cys(Hmbon), which can later be fully deprotected by TFA to generate free Cys. The utility of Cys(Hmboff/on) is demonstrated by the chemical synthesis of an erythropoietin segment, EPO[Cys98–Arg166]-OH through native chemical ligation.
Co-reporter:Yiming Li, Maiyun Yang, Yichao Huang, Xiaoda Song, Lei Liu and Peng R. Chen
Chemical Science (2010-Present) 2012 - vol. 3(Issue 9) pp:NaN2770-2770
Publication Date(Web):2012/06/19
DOI:10.1039/C2SC20433A
A series of alkene-bearing pyrrolysine analogues were synthesized and subsequently incorporated into proteins at two sites by a mutant PylRS–tRNA pair with excellent efficiency. This strategy allowed the site-specific labeling of proteins carrying single or double genetically encoded alkene handles via bioorthogonal thiol–ene ligation reactions.
Co-reporter:Shan Tang, Zhengpeng Wan, Yiren Gao, Ji-Shen Zheng, Jing Wang, Yan-Yan Si, Xin Chen, Hai Qi, Lei Liu and Wanli Liu
Chemical Science (2010-Present) 2016 - vol. 7(Issue 3) pp:NaN1895-1895
Publication Date(Web):2015/12/11
DOI:10.1039/C5SC03404C
We report the chemical synthesis of the first photo-activatable protein antigen that can be used to study antigen–antibody interaction mediated responses in B cells. This strategy facilitated fine tuning of the caged protein antigen to optimize its bioactivity and photochemical properties. One optimal molecule, HEL-K96NPE, was totally inert to hen egg lysozyme (HEL)-specific B cells and could only restore its antigenicity upon photoactivation. Combined with real time live cell imaging, the utility of HEL-K96NPE was demonstrated as a proof of concept to quantify B cell synapse formation and calcium influx responses at the single cell level.
Co-reporter:Jun Xu, Dong-Fen Luo, Bin Xiao, Zhao-Jing Liu, Tian-Jun Gong, Yao Fu and Lei Liu
Chemical Communications 2011 - vol. 47(Issue 14) pp:NaN4302-4302
Publication Date(Web):2011/03/07
DOI:10.1039/C1CC10359H
A copper-catalyzed process for trifluoromethylation of aryl, heteroaryl, and vinyl boronic acids has been developed. The reaction is conducted under mild conditions and shows tolerance to moisture and a variety of functional groups.
Co-reporter:Yi-Chao Huang, Cheng Cao, Xiang-Long Tan, Xiaoyu Li and Lei Liu
Inorganic Chemistry Frontiers 2014 - vol. 1(Issue 9) pp:NaN1054-1054
Publication Date(Web):2014/09/05
DOI:10.1039/C4QO00217B
PNA–peptide conjugates are useful molecular tools in chemical biology and biotechnology. Although several approaches have been developed to synthesize PNA–peptide conjugates, more efficient methods are still needed. In this report a new pNZ (p-nitrobenzyloxycarbonyl)/bis-Boc strategy was developed as an alternative backbone/nucleobase protecting group method. The mild deprotection conditions of pNZ group and pNZ's full orthogonality with Fmoc solid-phase synthesis enable a new dimension of synthetic flexibility and practicality to generate versatile PNA–peptide conjugates.



![2-(7-CHLORO-1,8-NAPHTHYRIDIN-2-YL)-3-[2-(1,4-DIOXA-8-AZASPIRO[4.5]DECAN-8-YL)-2-OXOETHYL]-3H-ISOINDOL-1-ONE](http://img.cochemist.com/ccimg/103300/103255-66-9.png)
![2-(7-CHLORO-1,8-NAPHTHYRIDIN-2-YL)-3-[2-(1,4-DIOXA-8-AZASPIRO[4.5]DECAN-8-YL)-2-OXOETHYL]-3H-ISOINDOL-1-ONE](http://img.cochemist.com/ccimg/103300/103255-66-9_b.png)


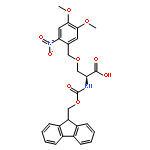
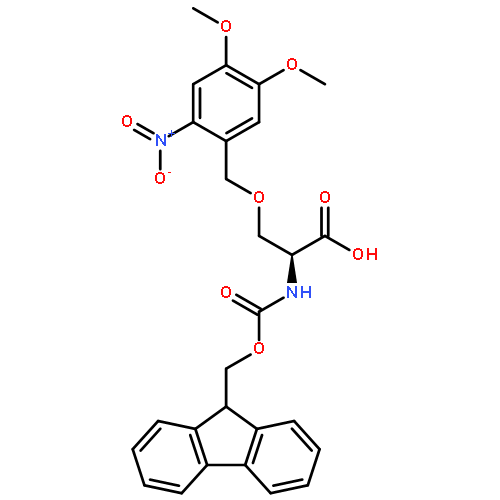




![L-Serine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 2-propen-1-yl ester](/data/chemimg/1685700/136497-85-3.png)
![L-Serine, N-[(9H-fluoren-9-ylmethoxy)carbonyl]-, 2-propen-1-yl ester](/data/chemimg/1685700/136497-85-3_b.png)






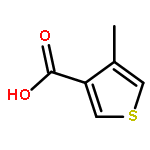





![2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/53700/53663-18-6.png)
![2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]benzoic acid](http://img.cochemist.com/ccimg/53700/53663-18-6_b.png)











![[1(3H)-ISOINDOLINONE-3-YL]ACETIC ACID](http://img.cochemist.com/ccimg/3900/3849-22-7.png)
![[1(3H)-ISOINDOLINONE-3-YL]ACETIC ACID](http://img.cochemist.com/ccimg/3900/3849-22-7_b.png)