Co-reporter:Ryan Morrow, Kartik Samanta, Tanusri Saha Dasgupta, Jie Xiong, John W. Freeland, Daniel Haskel, and Patrick M. Woodward
Chemistry of Materials 2016 Volume 28(Issue 11) pp:3666
Publication Date(Web):May 13, 2016
DOI:10.1021/acs.chemmater.6b00254
In order to rationalize and predict the behavior of compounds containing 5d transition metal ions, an understanding of the local moments and superexchange interactions from which their magnetic properties are derived is necessary. The magnetic and electrical properties of the ferrimagnetic double perovskites Ca2CoOsO6 and Ca2NiOsO6 studied here provide critical insight toward that goal. First-principles density functional theory (DFT) calculations indicate, and experimental measurements confirm, that the Os(VI) moments are directed antiparallel to the Co/Ni moments. X-ray magnetic circular dichroism (XMCD) measurements reveal that the orbital moment on osmium has a magnitude that is approximately 30% of the spin moment, and the two contributions oppose each other. Both the size and direction of the orbital moment are confirmed by the DFT calculations. The size of the Os(VI) total moment is predicted to be 0.6–0.7 μB by DFT calculations. The ferrimagnetic ground state is stabilized by strong antiferromagnetic coupling between the d2 Os(VI) ion and the d8/d7 Ni(II)/Co(II) ion. Not only does the observation of antiferromagnetic coupling violate the Goodenough–Kanamori rules, but also it is unusual in that it becomes stronger as the Os–O–Co/Ni bond angle decreases. This unusual behavior is shown to arise predominantly from coupling between Os t2g orbitals and Ni/Co eg orbitals, mediated by the intervening oxide ion. We further find that both compounds are spin–orbit assisted Mott insulators.
Co-reporter:Andrew R. Sharits, Jason F. Khoury, and Patrick M. Woodward
Inorganic Chemistry 2016 Volume 55(Issue 23) pp:12383-12390
Publication Date(Web):November 17, 2016
DOI:10.1021/acs.inorgchem.6b02295
Three doubly ordered double perovskites NaREMgWO6 (RE = La, Gd, Y) have been synthesized via traditional solid-state methods, doped with Eu3+, and characterized to evaluate their promise as Eu3+ phosphor hosts. NaYMgWO6, a new member of the family, was found to crystallize in the P21 space group and is isostructural with NaGdMgWO6. Emissions characteristic of Eu3+ ions (5D0 → 7F4,3,2,1,0) were observed, with the most intense transition being the 5D0 → 7F2 transition near 615 nm. Substitution of Eu3+ onto a more compressed RE site in the NaY1–xEuxMgWO6 and NaGd1–xEuxMgWO6 hosts results in a blue shift of the charge-transfer excitation band and an increase in the intensity of the 5D0 → 7F2 transition compared to NaLa1–xEuxMgWO6. All of the hosts can incorporate high concentrations of Eu3+ before concentration quenching is observed. When the rare-earth ion is either Gd3+ or Y3+, good energetic overlap between the Eu3+ charge-transfer band and the absorption of the host lattice results in sensitization and energy transfer from the perovskite host lattice to the Eu3+ activator sites. These hosts display comparable if not better luminescence than Y2O3:Eu3+, a commonly used commercial standard, demonstrating their promise as red phosphors.
Co-reporter:Spencer H. Porter, Jie Xiong, Maxim Avdeev, David Merz, Patrick M. Woodward, and Zhenguo Huang
Inorganic Chemistry 2016 Volume 55(Issue 12) pp:5772
Publication Date(Web):May 26, 2016
DOI:10.1021/acs.inorgchem.5b02755
Combined synchrotron and neutron powder diffraction indicates that A3V4(PO4)6 (A = Mg, Mn, Fe, Co, Ni) compounds crystallize with triclinic P1̅ symmetry. Lattice parameters expand as expected with successive increases in the ionic radius of the A2+ ion. Cation disorder on the octahedral sites increases as the ionic radii of A2+ ion decreases. Direct-current magnetic susceptibility measurements indicate that all compounds with magnetic A2+ ions order anti-ferromagnetically with transition temperatures ranging from 12 to 15 K. Effective magnetic moments for A3V4(PO4)6 (A = Mg, Mn, Fe, Co, Ni) are 5.16, 11.04, 10.08, 9.76, and 7.96 μB per formula unit, respectively, in line with calculated values for high-spin transition metal ions. With the exception of Co3V4(PO4)6 the ultraviolet–visible spectra are dominated by d–d transitions of the V3+ ions. The striking emerald green color of Co3V4(PO4)6 arises from the combined effects of d–d transitions involving both V3+ and Co2+.
Co-reporter:Spencer H. Porter, Zhenguo Huang, Shixue Dou, Samantha Brown-Xu, A.T.M. Golam Sarwar, Roberto C. Myers, and Patrick M. Woodward
Chemistry of Materials 2015 Volume 27(Issue 7) pp:2414
Publication Date(Web):February 26, 2015
DOI:10.1021/cm5044599
Three perovskite nitride oxides CeTiNO2, PrTiNO2, and NdTiNO2 have been synthesized, and their electronic structures and photocatalytic activities characterized and compared to LaTiNO2. All three compounds have band gaps that fall in the range of 2.0–2.1 eV, very similar to LaTiNO2, which enables them to absorb a significant fraction of the visible spectrum. Photocatalytic oxygen evolution studies under visible light irradiation in the presence of a sacrificial electron acceptor (Ag+) show that the activity of NdTiNO2 (16 μmol/g/h) is comparable to that of LaTiNO2 (17 μmol/g/h), while PrTiNO2 (11 μmol/g/h) and CeTiNO2 (5 μmol/g/h) have activities that are only 65% and 30% that of LaTiNO2. X-ray photoelectron spectroscopy measurements reveal the presence of partially occupied f-orbital states that lie in the band gap for CeTiNO2 and near the valence band maximum for PrTiNO2. As evidenced by time-resolved IR kinetic decay, these localized f-orbital states act as electron–hole recombination centers that inhibit the photocatalytic activities of both compounds. On the other hand, NdTiNO2, where the f-orbital energies fall below the valence band maximum, does not suffer from this effect.
Co-reporter:Ryan Morrow, John W. Freeland, and Patrick M. Woodward
Inorganic Chemistry 2014 Volume 53(Issue 15) pp:7983-7992
Publication Date(Web):July 15, 2014
DOI:10.1021/ic5006715
The synthesis, structure, and properties of the ordered double perovskites Sr2FeOsO6, Ca2FeOsO6, and SrCaFeOsO6 are reported. The latter two compounds have monoclinic P21/n symmetry and a–a–b+ tilting of the octahedra, while Sr2FeOsO6 is tetragonal with I4/m symmetry and a0a0c– tilting. Magnetic measurements indicate and neutron powder diffraction studies confirm that Ca2FeOsO6 is a ferrimagnet with a Curie temperature of 350 K. The ferrimagnetism is retained if half of the Ca2+ ions are replaced with larger Sr2+ ions to form SrCaFeOsO6 (TC = 210 K). This substitution reduces the degree of octahedral tilting, but unlike most perovskites, the magnetic ordering temperature decreases as the Fe–O–Os bond angles approach a linear geometry. In contrast, Sr2FeOsO6 orders antiferromagnetically, as previously reported. X-ray absorption spectroscopy confirms the assignment of Fe(III) and Os(V) oxidation states for all three compounds. In these insulating double perovskites, the magnetic ground state is governed by a competition between the four-bond Fe–O–Os–O–Fe antiferromagnetic superexchange coupling of Fe(III) ions and the two-bond Fe–O–Os antiferromagnetic superexchange coupling between neighboring Fe(III) and Os(V) ions. When the Fe–O–Os bonds are linear, as they are in the c direction in Sr2FeOsO6, the four-bond coupling between Fe(III) ions prevails. The competition shifts in favor of antiferromagnetic coupling of Fe(III) and Os(V) as the Fe–O–Os bond angles bend in response to chemical pressure.
Co-reporter:Hiroshi Mizoguchi, Nattamai S. P. Bhuvanesh, Young-Il Kim, Satoshi Ohara, and Patrick M. Woodward
Inorganic Chemistry 2014 Volume 53(Issue 19) pp:10570-10577
Publication Date(Web):September 10, 2014
DOI:10.1021/ic5016252
Colorless single crystals of LiSb(OH)6, SrSn(OH)6, and BaSn(OH)6, which are useful as precursors for the synthesis of LiSbO3, SrSnO3, and BaSnO3, were synthesized by a low-temperature hydrothermal method using a Teflon-lined autoclave at 380 K. The crystal structures were determined by single-crystal X-ray diffraction measurements. LiSb(OH)6 crystallizes in the trigonal space group P3̅1m with a = 5.3812(3)A, c = 9.8195(7)A, V = 246.25(3)A3, Z = 2. In this layered structure, [Li2Sb(OH)6]+ and [Sb(OH)6]− layers are alternately stacked along the c-direction. The [Li2Sb(OH)6]+ layer can be regarded as a cation-ordered CdCl2 layer. The [Sb(OH)6)]− layer is built up from isolated [Sb(OH)6]− octahedra, which are linked to each other via hydrogen bonding within the layer. BaSn(OH)6 and SrSn(OH)6 crystallize with monoclinic P21/n space group symmetry. The monoclinic structure possesses a CsCl-type packing of Ba2+/Sr2+ cations and [Sn(OH)6]2– anions. The [Sn(OH)6]2– polyhedra are connected to each other through hydrogen bonding to form a three-dimensional framework. The factors that favor these hitherto unknown crystal structures are discussed using a structure map that compares various M(OH)3 and M′M″(OH)6 compounds.
Co-reporter:Swagata Dey, Rebecca A. Ricciardo, Heather L. Cuthbert, and Patrick M. Woodward
Inorganic Chemistry 2014 Volume 53(Issue 9) pp:4394-4399
Publication Date(Web):April 22, 2014
DOI:10.1021/ic4031798
Using a combination of UV–visible spectroscopy and electronic structure calculations, we have characterized the electronic structures and optical properties of AWO4 (A = Mn, Co, Ni, Cu, Zn, or Mg) tungstates with the wolframite structure. In MgWO4 and ZnWO4, the lowest energy optical excitation is a ligand to metal charge transfer (LMCT) excitation from oxygen 2p nonbonding orbitals to antibonding W 5d orbitals. The energy of the LMCT transition in these two compounds is 3.95 eV for ZnWO4 and 4.06 eV for MgWO4. The charge transfer energies observed for the other compounds are significantly smaller, falling in the visible region of the spectrum and ranging from 2.3 to 3.0 eV. In these compounds, the partially occupied 3d orbitals of the A2+ ion act as the HOMO, rather than the O 2p orbitals. The lowest energy charge transfer excitation now becomes a metal-to-metal charge transfer (MMCT) excitation, where an electron is transferred from the occupied 3d orbitals of the A2+ ion to unoccupied antibonding W 5d states. The MMCT value for CuWO4 of 2.31 eV is the lowest in this series due to distortions of the crystal structure driven by the d9 configuration of the Cu2+ ion that lower the crystal symmetry to triclinic. The results of this study have important implications for the application of these and related materials as photocatalysts, photoanodes, pigments, and phosphors.
Co-reporter:Spencer H. Porter, Zhenguo Huang, and Patrick M. Woodward
Crystal Growth & Design 2014 Volume 14(Issue 1) pp:117-125
Publication Date(Web):October 31, 2013
DOI:10.1021/cg401230a
Symmetries and model structures are given for ABN2O (and ABNO2) perovskites that possess long-range ordering of anions in combination with a0a0c–, a–b0a–, and a–b+a– octahedral tilting. The stabilities of competing structures have been evaluated using density functional theory (DFT) calculations, which show that cis-ordered models are more stable than competing trans-ordered polymorphs. To test the validity of these predictions, the perovskite nitride oxides LaTaN2O, CeTaN2O, and PrTaN2O have been synthesized and characterized using neutron powder diffraction. CeTaN2O and PrTaN2O crystallize with orthorhombic Pnma symmetry (Ce: a = 5.69666(8), b = 8.03272(9), and c = 5.70893(7) Å; Pr: a = 5.6868(1), b = 8.0153(1), and c = 5.68057(8) Å) as a result of a–b+a– tilting of the octahedra. The structure of LaTaN2O is re-examined and found to possess orthorhombic Imma symmetry (a = 5.7093(1), b = 8.0591(2), and c = 5.7386(2) Å) as a result of a–b0a– tilting. No evidence for long-range anion order is found in any of the three compounds. Optical band gaps for these compounds are measured to be 2.0 eV (LaTaN2O), 1.9 eV (CeTaN2O), and 2.0 eV (PrTaN2O). These values are 0.6–0.7 eV smaller than CaTaNO2 where the Ta-centered octahedra tilt by a similar amount. As the nitrogen content increases, there is an increase in the overlap of the anion 2p orbitals, which increases the energy of the valence band maximum and narrows the band gap.
Co-reporter:Ryan Morrow ; Rohan Mishra ; Oscar D. Restrepo ; Molly R. Ball ; Wolfgang Windl ; Sabine Wurmehl ; Ulrike Stockert ; Bernd Büchner
Journal of the American Chemical Society 2013 Volume 135(Issue 50) pp:18824-18830
Publication Date(Web):November 18, 2013
DOI:10.1021/ja407342w
The insulating, fully ordered, double perovskite Sr2CoOsO6 undergoes two magnetic phase transitions. The Os(VI) ions order antiferromagnetically with a propagation vector k = (1/2, 1/2, 0) below TN1 = 108 K, while the high-spin Co(II) ions order antiferromagnetically with a propagation vector k = (1/2, 0, 1/2) below TN2 = 70 K. Ordering of the Os(VI) spins is accompanied by a structural distortion from tetragonal I4/m symmetry to monoclinic I2/m symmetry, which reduces the frustration of the face centered cubic lattice of Os(VI) ions. Density functional theory calculations show that the long-range Os–O–Co–O–Os and Co–O–Os–O–Co superexchange interactions are considerably stronger than the shorter Os–O–Co interactions. The poor energetic overlap between the 3d orbitals of Co and the 5d orbitals of Os appears to be responsible for this unusual inversion in the strength of short and long-range superexchange interactions.
Co-reporter:Hiroshi Mizoguchi, Ping Chen, Punit Boolchand, Vadim Ksenofontov, Claudia Felser, Paris W. Barnes, and Patrick M. Woodward
Chemistry of Materials 2013 Volume 25(Issue 19) pp:3858
Publication Date(Web):August 29, 2013
DOI:10.1021/cm4019309
The electronic and optical properties of the cubic perovskite, BaSnO3, are compared with the well-known transparent conducting oxides (TCOs), SnO2 and In2O3. The optical band gaps of the undoped compounds, as measured by diffuse reflectance spectroscopy on powdered samples, are 3.1 eV for BaSnO3 and 3.8 eV for SnO2. Electronic structure calculations show that both compounds possess a large conduction band dispersion, which suggests that BaSnO3, like SnO2, should be a good TCO if it can be n-doped. To explore this possibility the properties of substitutionally doped, BaSn1–xSbxO3 samples were investigated. The electrical conductivity increases drastically for BaSn1–xSbxO3 samples with x ≤ 0.05, showing a transition from an insulating to a metallic state. For higher doping levels, 0.05 < x < 0.15, the conductivity saturates at 4 S cm–1. This is accompanied by saturation in the expansion of the cubic lattice parameter. The color of the pellets changed from white (x = 0) to bluish black (x = 0.15). This darkening originates from the formation of an intense and broad optical absorption band centered at 1200–1300 nm for the more highly doped samples. This absorption band spans both the visible and the near-infrared regions, resulting in a loss of transparency. Various spectroscopic techniques were used to elucidate the observed behavior. Mössbauer spectroscopy reveals the presence of mixed valent antimony, Sb3+(5s2)/Sb5+(5s0), as x increases. ESR spectra collected on a BaSn0.99Sb0.01O3 sample indicate that only ∼5% of unpaired electrons are present as delocalized carriers. The other 95% are localized to form a Sn3+ trapped electron center. To explain the tendency for carrier localization in doped BaSnO3 a mechanism based on strong electron–phonon interactions in perovskites containing cations with (n – 1)d10ns0 electronic configuration is proposed. This mechanism explains why BaSn1–xSbxO3 possesses relatively poor TCO characteristics, in sharp contrast to widely used TCO materials Sn1–xSbxO2 and In1–xSnxO1.5.Keywords: mixed valence compounds; Mössbauer spectroscopy; perovskite oxides; transparent conductive oxides;
Co-reporter:Murthi S. Kandanapitiye, Benjamin Valley, Liu D. Yang, Allyson M. Fry, Patrick M. Woodward, and Songping D. Huang
Inorganic Chemistry 2013 Volume 52(Issue 6) pp:2790-2792
Publication Date(Web):March 6, 2013
DOI:10.1021/ic302262g
The gallium analogue of the soluble Prussian blue with the formula KGa[Fe(CN)6]·nH2O is synthesized and structurally characterized. A simple aqueous synthetic procedure for preparing nanoparticles of this novel coordination polymer is reported. The stability, in vitro ion exchange with ferrous ions, cytotoxicity, and cellular uptake of such nanoparticles coated with poly(vinylpyrrolidone) are investigated for potential applications of delivering Ga3+ ions into cells or removing iron from cells.
Co-reporter:Allyson M. Fry and Patrick M. Woodward
Crystal Growth & Design 2013 Volume 13(Issue 12) pp:5404-5410
Publication Date(Web):October 29, 2013
DOI:10.1021/cg401342q
The room temperature crystal structures of α-K3MoO3F3 and α-Rb3MoO3F3 have been solved via combined Rietveld refinements of synchrotron and neutron powder diffraction data. These two compounds are part of a broader family of A2BMO3F3 compounds that have been studied for their dielectric properties, but until now the complex crystal structures of the ferroelectric phases of these compounds were not known. At room temperature and below, these two isostructural compounds are tetragonal with I41 space group symmetry and unit cell parameters of a = 19.38613(3) Å, c = 34.86739(8) Å for α-K3MoO3F3 and a = 20.0748(4) Å, c = 36.1694(1) Å for α-Rb3MoO3F3. Their structures are related to the cubic double perovskite structure but are considerably more complicated due to noncooperative octahedral tilting and long-range orientational ordering of the polar MoO3F33– units. The pattern of octahedral tilting is equivalent to that seen in the α-K3AlF6 structure, which has I41/a symmetry, but orientational ordering of MoO3F33– units lowers the symmetry to I41. The polar space group symmetry is consistent with earlier reports of ferroelectricity in these compounds. Hence orientational ordering of the MoO3F33– units is directly responsible for the ferroelectric behavior.
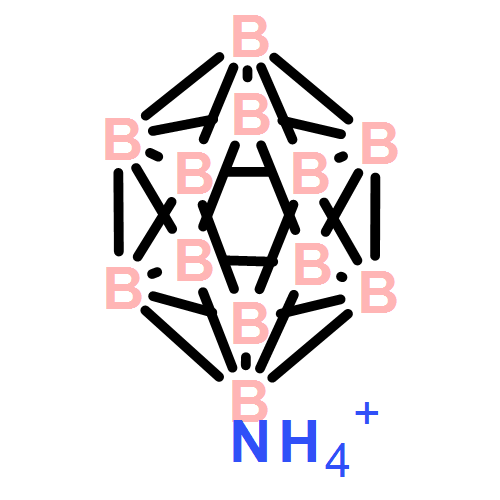
Co-reporter:Teshome B. Yisgedu, Zhenguo Huang, Xuenian Chen, Hima K. Lingam, Graham King, Aaron Highley, Sean Maharrey, Patrick M. Woodward, Richard Behrens, Sheldon G. Shore, Ji-Cheng Zhao
International Journal of Hydrogen Energy 2012 Volume 37(Issue 5) pp:4267-4273
Publication Date(Web):March 2012
DOI:10.1016/j.ijhydene.2011.11.112
The structure of (NH4)2B10H10 (1) was determined through powder XRD analysis. The thermal decomposition of 1 and (NH4)2B12H12 (2) was examined between 20 and 1000 °C using STMBMS methods. Between 200 and 400 °C a mixture of NH3 and H2 evolves from both compounds; above 400 °C only H2 evolves. The dihydrogen bonding interaction in 1 is much stronger than that in 2. The stronger dihydrogen bond in 1 resulted in a significant reduction by up to 60 °C, but with a corresponding 25% decrease in the yield of H2 in the lower temperature region and a doubling of the yield of NH3. The decomposition of 1 follows a lower temperature exothermic reaction pathway that yields substantially more NH3 than the higher temperature endothermic pathway of 2. Heating of 1 at 250 °C resulted in partial conversion of B10H102− to B12H122−. Both 1 and 2 form an insoluble polymeric material after decomposition. The elements of the reaction network that control the release of H2 from the B10H102− can be altered by conducting the experiment under conditions in which pressures of NH3 and H2 are either near, or away from, their equilibrium values.Highlights► The structure of (NH4)2B10H10 was determined for the first time. ► Stronger dihydrogen bonding in (NH4)2B10H10 compared to (NH4)2B12H12 resulted in a lower temperature H2/NH3 release. ► (NH4)2B10H10 thermally decomposed in an exothermic fashion compared to (NH4)2B12H12 which did in endothermic one. ► At 250 °C (NH4)2B10H10 was partially converted to [B12H12]2−.
Co-reporter:Allyson M. Fry ; Harry A. Seibel ; II; Indunil N. Lokuhewa
Journal of the American Chemical Society 2011 Volume 134(Issue 5) pp:2621-2625
Publication Date(Web):December 20, 2011
DOI:10.1021/ja208587e
Na1.5Ag1.5MoO3F3 and Na1.5Ag1.5WO3F3 have been synthesized by solid state reactions and structurally characterized using synchrotron X-ray and neutron powder diffraction. Unlike the vast majority of salts containing [MO3F3]3– anions (M = Mo, W) the oxyfluoride groups in Na1.5Ag1.5MoO3F3 and Na1.5Ag1.5WO3F3 are orientationally ordered, so that the Na+ ions are coordinated by fluorine and the Ag+ ions by oxygen. The resulting structure type, which has not previously been reported, is related to the LiNbO3 structure, but the combination of Na/Ag ordering and orientational ordering of the [MO3F3]3– anions produces a supercell that doubles the c-axis and changes the space group symmetry from R3 to R3̅. The use of hard (Na+) and soft (Ag+) cations to direct the orientational ordering of polar oxyfluoride building units provides a new approach to the design of polar materials.
Co-reporter:Susana Garcia-Martin, Graham King, Esteban Urones-Garrote, Gwilherm Nénert, and Patrick M. Woodward
Chemistry of Materials 2011 Volume 23(Issue 2) pp:163
Publication Date(Web):December 17, 2010
DOI:10.1021/cm102592p
Electron, neutron, and synchrotron X-ray diffraction together with transmission electron microscopy studies reveal the spontaneous formation of a complex superlattice in bulk samples of the perovskite KLaMnWO6. The superlattice structure, which possesses P4̅2m space group symmetry with a = 40.0637(7) Å and c = 8.1306(3) Å, results from a two-dimensional compositional modulation of the A-site cations (K+ and La3+), combined with a complex pattern of tilts involving the corner connected octahedra. The basic pattern of octahedral tilting involves out-of-phase tilts of neighboring octahedra about the pseudocubic a and b axes (a−a−c0 tilting). Unexpectedly, the out-of-phase tilting is disrupted in both directions by an in-phase tilt once every five octahedra. The occurrence of regularly repeating, well-separated in-phase tilts helps to alleviate strains that arise from formation of the compositionally modulated chessboard superlattice.
Co-reporter:Graham King and Patrick M. Woodward
Journal of Materials Chemistry A 2010 vol. 20(Issue 28) pp:5785-5796
Publication Date(Web):15 Apr 2010
DOI:10.1039/B926757C
Although both A- and B-site cations have the same simple cubic topology in the perovskite structure they typically adopt different patterns of chemical order. As a general rule B-site cations order more readily than A-site cations. When cation ordering does occur, rock salt ordering of B/B′ cations is favored in A2BB′X6 perovskites, whereas layered ordering of A/A′ cations is favored in AA′B2X6 and AA′BB′X6 perovskites. The unexpected tendency for A-site cations to order into layers stems from the bond strains that would result at the anion site if A and A′ cations of different size were to order with a rock salt arrangement. The bonding instabilities that are created by layered ordering are generally offset either by anion vacancies or second order Jahn–Teller distortions of a B-site cation. Novel types of A-site cation ordering can be stabilized by a+a+a+ or a+a+c− tilting of the octahedra.
Co-reporter:Harry A. Seibel II, Pavel Karen, Timothy R. Wagner and Patrick M. Woodward
Journal of Materials Chemistry A 2009 vol. 19(Issue 4) pp:471-477
Publication Date(Web):03 Dec 2008
DOI:10.1039/B815758H
Reaction pathways to nitrogen- and fluorine-doped TiO2 have been investigated and the compositions analyzed by neutron powder diffraction, nitrogen analyses and titrations of Ti3+. The reported formation of TiNF under pyrolysis of (NH4)2TiF6 in NH3 could not be reproduced when H2O was carefully excluded. If special precautions are not taken to exclude H2O, a product of the previously reported olive-green appearance is obtained; TiN0.05O1.89F0.06, in which 1% of Ti is trivalent. The prolonged hydrolysis and ammonolysis leading to this product proceeds via the intermediates (NH4)1−xTiOF3−x, which adopts the hexagonal tungsten–bronze structure, and/or TiOF2. The latter is therefore suggested as a convenient starting material for doped anatase pigments. Reflectance measurements couple the green color with valence to conduction band excitations (Eg = 2.3 eV) and intraband transitions involving Ti3+. The green phase can be converted to a brilliant yellow in the presence of water vapor at 400–600 °C, which further hydrolyzes the fluoride and oxidizes the titanium, yielding TiN0.04O1.92F0.04 in a particular case. The yellow color and band gap (Eg = 2.4 eV) may be promising for applications as a pigment or photocatalyst. Neutron powder diffraction characterizations and UV–Visible reflectance measurements indicate homogenous doping throughout the bulk. The combined results suggest that the green and yellow phases are part of a homogeneity range adjacent to TiO2, in which the band gap narrowing results largely from nitrogen for oxygen subsitution and the green color is linked to formation of Ti3+ defects.
Co-reporter:Michelle R. Dolgos, Alexandra M. Paraskos, Matthew W. Stoltzfus, Samantha C. Yarnell, Patrick M. Woodward
Journal of Solid State Chemistry 2009 Volume 182(Issue 7) pp:1964-1971
Publication Date(Web):July 2009
DOI:10.1016/j.jssc.2009.04.032
The electronic structures of six ternary metal oxides containing isolated vanadate ions, Ba3(VO4)2, Pb3(VO4)2, YVO4, BiVO4, CeVO4 and Ag3VO4 were studied using diffuse reflectance spectroscopy and electronic structure calculations. While the electronic structure near the Fermi level originates largely from the molecular orbitals of the vanadate ion, both experiment and theory show that the cation can strongly influence these electronic states. The observation that Ba3(VO4)2 and YVO4 have similar band gaps, both 3.8 eV, shows that cations with a noble gas configuration have little impact on the electronic structure. Band structure calculations support this hypothesis. In Pb3(VO4)2 and BiVO4 the band gap is reduced by 0.9–1.0 eV through interactions of (a) the filled cation 6s orbitals with nonbonding O 2p states at the top of the valence band, and (b) overlap of empty 6p orbitals with antibonding V 3d–O 2p states at the bottom of the conduction band. In Ag3VO4 mixing between filled Ag 4d and O 2p states destabilizes states at the top of the valence band leading to a large decrease in the band gap (Eg=2.2 eV). In CeVO4 excitations from partially filled 4f orbitals into the conduction band lower the effective band gap to 1.8 eV. In the Ce1−xBixVO4 (0≤x≤0.5) and Ce1−xYxVO4 (x=0.1, 0.2) solid solutions the band gap narrows slightly when Bi3+ or Y3+ are introduced. The nonlinear response of the band gap to changes in composition is a result of the localized nature of the Ce 4f orbitals.The electronic structures of six vanadate salts, Ba3(VO4)2, Pb3(VO4)2, YVO4, BiVO4, Ag3VO4 and CeVO4, are studied. The results show that the oxygen to vanadium charge transfer, which is largely responsible for the electronic structure near the Fermi level, can be altered significantly through interactions with the surrounding cations.
Co-reporter:R.A. Ricciardo, A.J. Hauser, F.Y. Yang, H. Kim, W. Lu, P.M. Woodward
Materials Research Bulletin 2009 44(2) pp: 239-247
Publication Date(Web):
DOI:10.1016/j.materresbull.2008.10.015
Co-reporter:Graham King, Srinivasa Thimmaiah, Akansha Dwivedi and Patrick M. Woodward
Chemistry of Materials 2007 Volume 19(Issue 26) pp:6451
Publication Date(Web):November 20, 2007
DOI:10.1021/cm0716708
Six new AA′BB′O6 perovskites KLaMnWO6, NaLaMnWO6, NaNdMnWO6, NaTbMnWO6, NaNdCoWO6, and NaNdMgWO6 have been prepared. Each possesses the unusual combination of layered ordering of the A-site cations and rock-salt ordering of the B-site cations. The structure and properties of these samples have been characterized using monochromatic X-ray and neutron powder diffraction, UV–vis diffuse reflectance spectroscopy, and SQUID magnetometry. NaLaMnWO6, NaNdMnWO6, and NaTbMnWO6 adopt a structure with monoclinic P21 symmetry arising from the combination of cation ordering and a–a–c+ octahedral tilting. The structures of the other three compounds are similar, but the presence of satellite reflections in the neutron diffraction data suggests a more complicated superstructure. Each of the four AA′MnWO6 samples shows a paramagnetic to antiferromagnetic transition with Néel temperatures ranging from 6 to 15 K. The NaTbMnWO6 compound shows a second magnetic transition at ∼9 K. The origin of two magnetic phase transitions appears to arise from coupling between the Mn2+ sublattice and the Tb3+ sublattice.
Co-reporter:Michael W Lufaso, Patrick M Woodward, Joshua Goldberger
Journal of Solid State Chemistry 2004 Volume 177(4–5) pp:1651-1659
Publication Date(Web):April–May 2004
DOI:10.1016/j.jssc.2003.12.020
Polycrystalline samples of A2MnMO6 (A=Sr, Ca; M=Nb, Sb, Ru) were prepared by conventional solid state synthesis and their crystal structures were determined using neutron powder diffraction data. All six compounds can be classified as distorted, disordered perovskites. The Mn3+/M5+ distribution is disordered in all six compounds. The strontium containing compounds, Sr2MnMO6 (M=Nb, Sb, Ru), undergo out of phase rotations of the octahedra about the c-axis (tilt system a0a0c−) leading to tetragonal I4/mcm space group symmetry. The calcium containing compounds, Ca2MnMO6 (M=Nb, Ru, Sb), have orthorhombic Pnma space group symmetry, as a result of a GdFeO3-type octahedral tilting distortion (tilt system a−b+a−). A cooperative Jahn–Teller distortion is observed in Sr2MnSbO6 and Sr2MnRuO6, but it is much smaller than the distortion observed in LnMnO3 (Ln=lanthanide ion) perovskites. It is possible that Jahn–Teller distortions of the MnO6 octahedra take place on a short-range length scale in the other four compounds, but there is little or no evidence for cooperative ordering of the local distortions. These findings demonstrate a link between orbital ordering, cation ordering and octahedral tilting.
Co-reporter:Hiroshi Mizoguchi, Nattamai S. P. Bhuvanesh and Patrick M. Woodward
Chemical Communications 2003 (Issue 9) pp:1084-1085
Publication Date(Web):07 Apr 2003
DOI:10.1039/B300635B
Characterization of polycrystalline samples of the trirutile oxides ZnBi2O6 and MgBi2O6 reveals temperature independent conductivity (0.4 and 0.01 S cm−1), a negative Seebeck coefficient (−0.035 and −0.025 mV K−1), and an optical band gap that falls at the low energy end of visible region (1.7 and 1.8 eV), this combination of attributes, indicating that these compounds are degenerate n-type semiconductors, has not previously been observed in a Bi5+ oxide.
Co-reporter:Hank W. Eng, Paris W. Barnes, Benjamin M. Auer, Patrick M. Woodward
Journal of Solid State Chemistry 2003 Volume 175(Issue 1) pp:94-109
Publication Date(Web):October 2003
DOI:10.1016/S0022-4596(03)00289-5
Computational and experimental studies using linear muffin tin orbital methods and UV-visible diffuse reflectance spectroscopy, respectively, were performed to quantitatively probe the relationships between composition, crystal structure and the electronic structure of oxides containing octahedrally coordinated d0 transition metal ions. The ions investigated in this study (Ti4+, Nb5+, Ta5+, Mo6+, and W6+) were examined primarily in perovskite and perovskite-related structures. In these compounds the top of the valence band is primarily oxygen 2p non-bonding in character, while the conduction band arises from the π∗ interaction between the transition metal t2g orbitals and oxygen. For isostructural compounds the band gap increases as the effective electronegativity of the transition metal ion decreases. The effective electronegativity decreases in the following order: Mo6+>W6+>Nb5+∼Ti4+>Ta5+. The band gap is also sensitive to changes in the conduction band width, which is maximized for structures possessing linear M–O–M bonds, such as the cubic perovskite structure. As this bond angle decreases (e.g., via octahedral tilting distortions) the conduction band narrows and the band gap increases. Decreasing the dimensionality from 3-D (e.g., the cubic perovskite structure) to 2-D (e.g., the K2NiF4 structure) does not significantly alter the band gap, whereas completely isolating the MO6 octahedra (e.g., the ordered double perovskite structure) narrows the conduction band width dramatically and leads to a significant increase in the band gap. Inductive effects due to the presence of electropositive “spectator” cations (alkali, alkaline earth, and rare-earth cations) tend to be small and can generally be neglected.
Co-reporter:Harry A. Seibel II, Pavel Karen, Timothy R. Wagner and Patrick M. Woodward
Journal of Materials Chemistry A 2009 - vol. 19(Issue 4) pp:NaN477-477
Publication Date(Web):2008/12/03
DOI:10.1039/B815758H
Reaction pathways to nitrogen- and fluorine-doped TiO2 have been investigated and the compositions analyzed by neutron powder diffraction, nitrogen analyses and titrations of Ti3+. The reported formation of TiNF under pyrolysis of (NH4)2TiF6 in NH3 could not be reproduced when H2O was carefully excluded. If special precautions are not taken to exclude H2O, a product of the previously reported olive-green appearance is obtained; TiN0.05O1.89F0.06, in which 1% of Ti is trivalent. The prolonged hydrolysis and ammonolysis leading to this product proceeds via the intermediates (NH4)1−xTiOF3−x, which adopts the hexagonal tungsten–bronze structure, and/or TiOF2. The latter is therefore suggested as a convenient starting material for doped anatase pigments. Reflectance measurements couple the green color with valence to conduction band excitations (Eg = 2.3 eV) and intraband transitions involving Ti3+. The green phase can be converted to a brilliant yellow in the presence of water vapor at 400–600 °C, which further hydrolyzes the fluoride and oxidizes the titanium, yielding TiN0.04O1.92F0.04 in a particular case. The yellow color and band gap (Eg = 2.4 eV) may be promising for applications as a pigment or photocatalyst. Neutron powder diffraction characterizations and UV–Visible reflectance measurements indicate homogenous doping throughout the bulk. The combined results suggest that the green and yellow phases are part of a homogeneity range adjacent to TiO2, in which the band gap narrowing results largely from nitrogen for oxygen subsitution and the green color is linked to formation of Ti3+ defects.
Co-reporter:Graham King and Patrick M. Woodward
Journal of Materials Chemistry A 2010 - vol. 20(Issue 28) pp:NaN5796-5796
Publication Date(Web):2010/04/15
DOI:10.1039/B926757C
Although both A- and B-site cations have the same simple cubic topology in the perovskite structure they typically adopt different patterns of chemical order. As a general rule B-site cations order more readily than A-site cations. When cation ordering does occur, rock salt ordering of B/B′ cations is favored in A2BB′X6 perovskites, whereas layered ordering of A/A′ cations is favored in AA′B2X6 and AA′BB′X6 perovskites. The unexpected tendency for A-site cations to order into layers stems from the bond strains that would result at the anion site if A and A′ cations of different size were to order with a rock salt arrangement. The bonding instabilities that are created by layered ordering are generally offset either by anion vacancies or second order Jahn–Teller distortions of a B-site cation. Novel types of A-site cation ordering can be stabilized by a+a+a+ or a+a+c− tilting of the octahedra.
.jpg)
























![37H,39H-Tetranaphtho[2,3-b:2',3'-g:2'',3''-l:2''',3'''-q]porphyrazine,5,9,14,18,23,27,32,36-octabutoxy-](/data/chemimg/114000/105528-25-4.png)
![37H,39H-Tetranaphtho[2,3-b:2',3'-g:2'',3''-l:2''',3'''-q]porphyrazine,5,9,14,18,23,27,32,36-octabutoxy-](/data/chemimg/114000/105528-25-4_b.png)