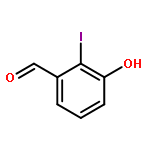
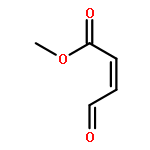
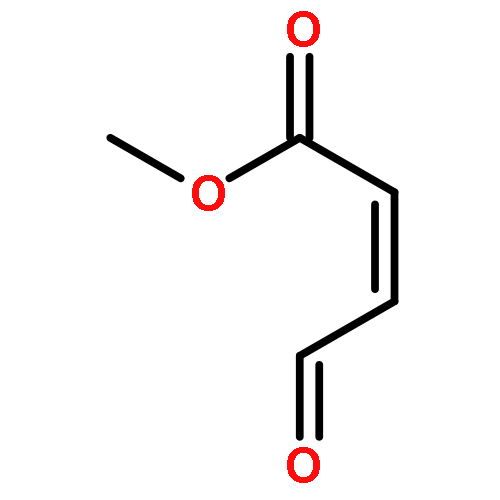
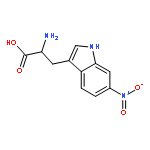
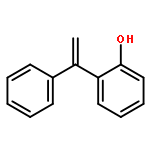
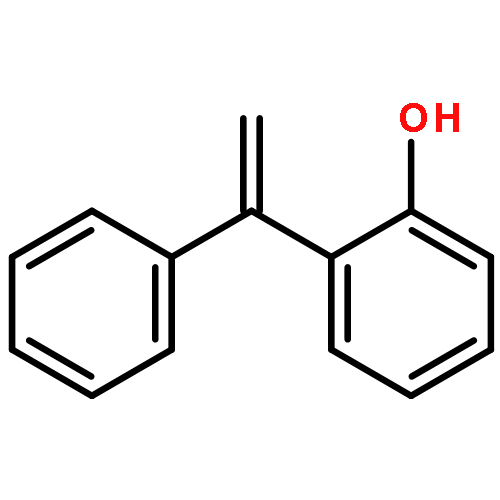
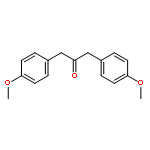
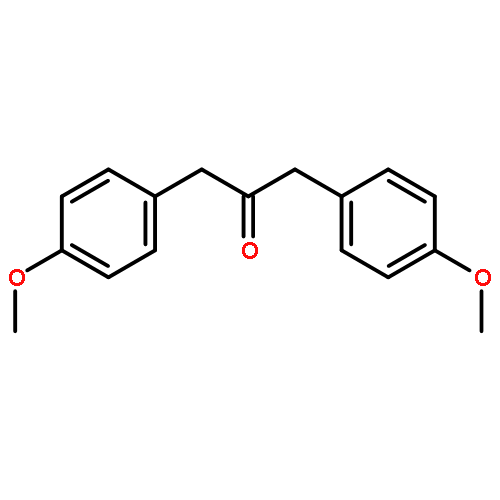
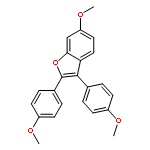
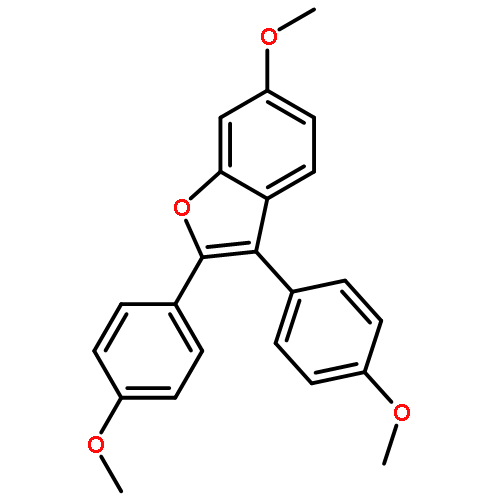
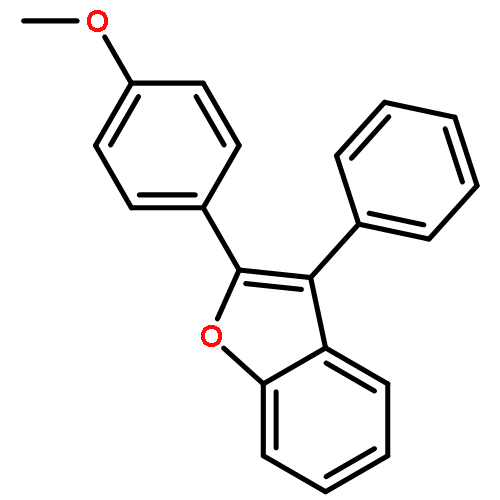
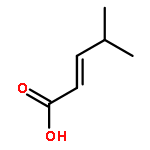
Co-reporter:Lei Li, Paruke Aibibula, Qianlan Jia, and Yanxing Jia
Organic Letters May 19, 2017 Volume 19(Issue 10) pp:
Publication Date(Web):May 3, 2017
DOI:10.1021/acs.orglett.7b00983
A divergent approach for the enantioselective total synthesis of eight monoterpenoid indole alkaloids was developed. The approach allows the first total syntheses of naucleamides A–C and E in only 6–8 steps and also enables the efficient synthesis of geissoschizine, geissoschizol, (E)-isositsirikine, and 16-epi-(E)-isositsirikine in 10–11 steps from commercially available crotonic aldehyde. The synthesis features a one-pot organocatalyzed asymmetric Michael addition/Pictet–Spengler reaction. Notably, biomimetic synthesis of naucleamide E was achieved by oxidative cyclization of naucleamide A.
Co-reporter:Haichao Liu, Xiwu Zhang, Dong Shan, Mallesham Pitchakuntla, Yongfan Ma, and Yanxing Jia
Organic Letters June 16, 2017 Volume 19(Issue 12) pp:
Publication Date(Web):June 8, 2017
DOI:10.1021/acs.orglett.7b01504
A new approach for the divergent total synthesis of eight ergot alkaloids is reported. The approach allows the first total syntheses of pyroclavine, pibocin A, 9-deacetoxyfumigaclavine C, and fumigaclavine G and also enables the efficient synthesis of festuclavine, costaclavine, epi-costaclavine, and dihydrosetoclavine. The main feature of the synthesis is the use of an unprecedented Pd-catalyzed intramolecular Larock indole annulation/Tsuji–Trost allylation cascade to assemble the tetracyclic core in one step.
Co-reporter:Lunyong Shi, Lingyu Li, Jun Wang, Bin Huang, Kewu Zeng, Hongwei Jin, Qingying Zhang, Yanxing Jia
Tetrahedron Letters 2017 Volume 58, Issue 20(Issue 20) pp:
Publication Date(Web):17 May 2017
DOI:10.1016/j.tetlet.2017.03.086
•The first total synthesis of similisines A, B and stereoisomers was accomplished.•The synthesis avoided any protecting-group manipulations.•The key spirocenter was constructed via intramolecular Friedel-Crafts cyclization.•The cytotoxic and NO inhibitory activities of synthetic compounds were discussed.The first total synthesis of similisines A and B, a pair of unprecedented polybrominated spiro-trisindole enantiomers isolated from Laurencia similis, and their all stereoisomers had been accomplished in 6 steps from the known 5,6-dibromoindole. The described synthesis avoided any protecting-group manipulations, and the key all-carbon spirocenters were constructed via an intramolecular Friedel-Crafts cyclization. In addition, the rotamers of similisines and cytotoxic and NO production inhibitory activities of synthetic compounds were also discussed.Download high-res image (104KB)Download full-size image
Co-reporter:Haichao Liu
Natural Product Reports (1984-Present) 2017 vol. 34(Issue 4) pp:411-432
Publication Date(Web):2017/04/05
DOI:10.1039/C6NP00110F
Covering: 2000 to 2017
Ergot alkaloids are among the most important pharmaceuticals and natural toxins. Significant progress has been achieved in recent years on the research of ergot alkaloids. In this review, we re-introduced the history of ergot alkaloids. Meanwhile, we summarized all the natural products and semi-synthetic derivatives of ergot alkaloids. We also briefly described the biosynthesis and semi-synthesis of ergot alkaloid drugs from raw materials obtained by fermentation. Moreover, we reviewed the advances that have been made in the total synthesis of ergot alkaloids since 2000.
Co-reporter:Xiwu Zhang, Haichao Liu and Yanxing Jia
Chemical Communications 2016 vol. 52(Issue 49) pp:7665-7667
Publication Date(Web):19 May 2016
DOI:10.1039/C6CC02600A
A concise and efficient synthetic route to 4,5-fused tricyclic 2-quinolones through the palladium-catalyzed carbonylative annulation of alkyne-tethered N-substituted o-iodoanilines has been developed. This reaction proceeds smoothly under mild reaction conditions and exhibits exceptional tolerance to a variety of functional groups. It has been successfully applied to the efficient synthesis of BI 224436, an HIV integrase inhibitor.
Co-reporter:Bin Huang; Fengying Zhang; Gang Yu; Yan Song; Xintong Wang; Meiliang Wang; Zehui Gong; Ruibin Su
Journal of Medicinal Chemistry 2016 Volume 59(Issue 8) pp:3953-3963
Publication Date(Web):March 29, 2016
DOI:10.1021/acs.jmedchem.6b00132
(−)-Incarvillateine (INCA) is the major antinociceptive component of Incarvillea sinensis, which has been used to treat rheumatism and relieve pain in traditional Chinese medicine. We have developed a concise and general synthetic approach for INCA, which enabled gram-scale asymmetric syntheses of (−)-INCA, (−)-incarvilline, (−)-isoincarvilline, and six other INCA analogues. The synthesis of isoincarvilline was reported for the first time. Three structurally simplified analogues of INCA were also synthesized. In vivo screening found that INCA and two structurally optimized analogues were efficacious in preventing the acetic acid-induced writhing response. Moreover, their analgesic efficacy was demonstrated in formalin induced pain model. More importantly, administration of 20 or 40 mg/kg INCA and two structurally optimized analogues showed strong analgesic effects in spared nerve injury (SNI) model, and their effective doses were lower than the current gold standard, gabapentin (100 mg/kg in this model).
Co-reporter:Pengyu Tao
Science China Chemistry 2016 Volume 59( Issue 9) pp:1109-1125
Publication Date(Web):2016 September
DOI:10.1007/s11426-016-0058-7
The transition metal-mediated C–H bond activation has emerged as a powerful and ideal method for the total syntheses of natural products and pharmaceuticals, and has had a significant impact on synthetic planning and strategy in complex natural products. In this review, we describe selected recent examples of the transition metal-mediated C–H bond activation strategies for the rapid syntheses of natural products.
Co-reporter:Fengying Zhang, Bin Wang, Pritesh Prasad, Robert J. Capon, and Yanxing Jia
Organic Letters 2015 Volume 17(Issue 6) pp:1529-1532
Publication Date(Web):March 2, 2015
DOI:10.1021/acs.orglett.5b00327
The first asymmetric total synthesis of the bis-indole marine alkaloid (+)-dragmacidin D (1) has been achieved. This synthesis revises an earlier configurational assignment based on biosynthetic considerations and assigns an R absolute configuration to (+)-1. The current studies reveal that natural dragmacidin D is isolated as either a racemate or a scalemic mixture (39% ee).
Co-reporter:Yinliang Guo, Qiang Liu and Yanxing Jia
Chemical Communications 2015 vol. 51(Issue 5) pp:889-891
Publication Date(Web):24 Nov 2014
DOI:10.1039/C3CC48897G
A concise synthesis of the tricyclic skeleton of crotobarin and crotogoudin via a gold-catalyzed 1,6-enyne cycloisomerization reaction is reported.
Co-reporter:Lei Li;Qiao Yang;Dr. Yuan Wang ; Yanxing Jia
Angewandte Chemie International Edition 2015 Volume 54( Issue 21) pp:6255-6259
Publication Date(Web):
DOI:10.1002/anie.201411338
Abstract
The catalytic asymmetric total syntheses of (−)-galanthamine (1) and (−)-lycoramine (2) have been achieved by using a conceptually new strategy featuring two metal-catalyzed reactions as the key steps. A new method for the construction of 3,4-fused benzofurans has been developed through a palladium-catalyzed intramolecular Larock annulation reaction, which was successfully applied to the construction of the ABD tricyclic skeleton of 1 and 2. To achieve the asymmetric synthesis of 1 and 2, a ScIII/N,N′-dioxide complex was used to catalyze the enantioselective conjugate addition of 3-alkyl-substituted benzofuranone to methyl vinyl ketone for the construction of a chiral quaternary carbon center.
Co-reporter:Zhuang Chen, Shiqiang Zhou, and Yanxing Jia
The Journal of Organic Chemistry 2015 Volume 80(Issue 24) pp:12545-12551
Publication Date(Web):November 30, 2015
DOI:10.1021/acs.joc.5b02402
The formal synthesis of (+)-kopsihainanine A has been achieved via stereoselective reduction of tetracyclic iminium ion intermediates (24). However, attempts to synthesize (+)-limaspermidine by reduction of the same tetracyclic iminium ion intermediates have failed. The synthesis features a Suzuki cross-coupling reaction, a cyclization reaction mediated by trifluoromethanesulfonic anhydride, and stereoselective reduction of an iminium ion.
Co-reporter:Bin Huang;Lei Guo; Yanxing Jia
Angewandte Chemie International Edition 2015 Volume 54( Issue 46) pp:13599-13603
Publication Date(Web):
DOI:10.1002/anie.201506575
Abstract
The first enantioselective synthesis of (−)-pallavicinin and (+)-neopallavicinin has been achieved in 15 steps. The described synthesis avoids protecting-group manipulations by synthesis designs predicated on highly chemo- and stereoselective transformations. Highlights of the synthesis include a palladium-catalyzed enantioselective decarboxylative allylation to form the chiral all-carbon quaternary stereocenter, a palladium-catalyzed oxidative cyclization to assemble the [3.2.1]-bicyclic moiety, and an unprecedented LiBHEt3-induced fragmentation/protonation of an α-hydroxy epoxide to form the α-furan ketone with the desired configuration.
Co-reporter:Bin Huang;Lei Guo; Yanxing Jia
Angewandte Chemie 2015 Volume 127( Issue 46) pp:13803-13807
Publication Date(Web):
DOI:10.1002/ange.201506575
Abstract
The first enantioselective synthesis of (−)-pallavicinin and (+)-neopallavicinin has been achieved in 15 steps. The described synthesis avoids protecting-group manipulations by synthesis designs predicated on highly chemo- and stereoselective transformations. Highlights of the synthesis include a palladium-catalyzed enantioselective decarboxylative allylation to form the chiral all-carbon quaternary stereocenter, a palladium-catalyzed oxidative cyclization to assemble the [3.2.1]-bicyclic moiety, and an unprecedented LiBHEt3-induced fragmentation/protonation of an α-hydroxy epoxide to form the α-furan ketone with the desired configuration.
Co-reporter:Lei Li;Qiao Yang;Dr. Yuan Wang ; Yanxing Jia
Angewandte Chemie 2015 Volume 127( Issue 21) pp:6353-6357
Publication Date(Web):
DOI:10.1002/ange.201411338
Abstract
The catalytic asymmetric total syntheses of (−)-galanthamine (1) and (−)-lycoramine (2) have been achieved by using a conceptually new strategy featuring two metal-catalyzed reactions as the key steps. A new method for the construction of 3,4-fused benzofurans has been developed through a palladium-catalyzed intramolecular Larock annulation reaction, which was successfully applied to the construction of the ABD tricyclic skeleton of 1 and 2. To achieve the asymmetric synthesis of 1 and 2, a ScIII/N,N′-dioxide complex was used to catalyze the enantioselective conjugate addition of 3-alkyl-substituted benzofuranone to methyl vinyl ketone for the construction of a chiral quaternary carbon center.
Co-reporter:Fengying Zhang, Lei Guo, Weimin Hu, Yanxing Jia
Tetrahedron 2015 Volume 71(Issue 22) pp:3756-3762
Publication Date(Web):3 June 2015
DOI:10.1016/j.tet.2014.06.095
A novel synthetic approach for the expedient synthesis of (−)-decursivine and analogues via two cascade sequences was developed. During this study, a new Pd(OAc)2-mediated Heck reaction/de-protection/oxidative cyclization cascade sequence was discovered unexpectedly. Other highlights of the synthesis include a cascade Witkop photocyclization/elimination sequence, and a Sm-mediated reduction of benzo[b]furan.
Co-reporter:Lei Guo, Fengying Zhang, Weimin Hu, Lei Li and Yanxing Jia
Chemical Communications 2014 vol. 50(Issue 25) pp:3299-3302
Publication Date(Web):07 Feb 2014
DOI:10.1039/C3CC49717H
A new palladium-catalyzed method for the synthesis of benzofurans by reaction of 2-hydroxystyrenes and iodobenzenes via a C–H activation/oxidation tandem reaction has been unprecedentedly discovered. By using this strategy, the overall synthetic efficiency of the synthesis of decursivine and its analogues was indeed improved. Preliminary mechanistic studies shed light into the possible mechanisms.
Co-reporter:Shiqiang Zhou and Yanxing Jia
Organic Letters 2014 Volume 16(Issue 12) pp:3416-3418
Publication Date(Web):June 2, 2014
DOI:10.1021/ol501341b
The total synthesis of (−)-goniomitine has been accomplished in 11 steps starting from commercially available diethyl l-malate. The synthesis features a chiral pool approach to prepare the chiral C-9 unit containing a quaternary carbon center, an Ir-catalyzed C–H borylation to synthesize the 2-indoleboronic acid pinacol ester, and a Suzuki reaction to couple together the two key intermediates. Notably, the high degree of convergence of this strategy makes it particularly amenable to the total synthesis of other aspidosperma family natural products.
Co-reporter:Pengyu Tao and Yanxing Jia
Chemical Communications 2014 vol. 50(Issue 55) pp:7367-7370
Publication Date(Web):21 May 2014
DOI:10.1039/C4CC02947J
The rhodium(III)-catalyzed intramolecular annulation of alkyne-tethered acetanilides for the synthesis of fused tricyclic indole scaffolds via C–H activation has been developed, which has the potential for the synthesis of many indole alkaloids. This reaction proceeds under mild reaction conditions and with tolerance to a variety of functional groups.
Co-reporter:Pengyu Tao;Jingjing Liang
European Journal of Organic Chemistry 2014 Volume 2014( Issue 26) pp:5735-5748
Publication Date(Web):
DOI:10.1002/ejoc.201402672
Abstract
A full account of the concise total syntheses of dictyodendrins B and E, and the formal synthesis of dictyodendrin C are described. A palladium-catalysed Larock indole synthesis was used to form the highly substituted indole core, and a palladium-mediated one-pot consecutive Buchwald–Hartwig amination/C–H activation reaction was used to construct the key pyrrolo[2,3-c]carbazole core. Unsuccessful synthetic strategies are also discussed.
Co-reporter:Bin Wang, Hua Qin, Fengying Zhang, Yanxing Jia
Tetrahedron Letters 2014 Volume 55(Issue 9) pp:1561-1563
Publication Date(Web):26 February 2014
DOI:10.1016/j.tetlet.2014.01.063
Synthesis of the pivotal cycloheptannelated indole fragment of indole alkaloid dragmacidin E is achieved. The synthesis features a Pd-catalyzed indole synthesis reaction for the preparation of 3,7-disubstituted indole, a regioselective intramolecular Friedel–Crafts reaction for the construction of the 3,4-bridged cycloheptannelated indole skeleton, and a coupling reaction of vinyl triflate with diorganocuprate.
Co-reporter:Yan Gao, Dong Shan, Yanxing Jia
Tetrahedron 2014 70(34) pp: 5136-5141
Publication Date(Web):
DOI:10.1016/j.tet.2014.05.108
Co-reporter:Lei Li, Weimin Hu, Yanxing Jia
Tetrahedron 2014 70(42) pp: 7753-7762
Publication Date(Web):
DOI:10.1016/j.tet.2014.05.082
Co-reporter:Qiang Liu, Qingjiang Li, Yongfan Ma, and Yanxing Jia
Organic Letters 2013 Volume 15(Issue 17) pp:4528-4531
Publication Date(Web):August 9, 2013
DOI:10.1021/ol4020877
The first Pd-catalyzed method for direct olefination at the C4 position of tryptophan derivatives has been developed via C–H activation to prepare 4-substituted tryptophans, which could be used for the synthesis of many hemiterpenoid indole alkaloids. This reaction proceeds under mild reaction conditions and with exceptional tolerance to a variety of functional groups. Furthermore, the efficiency of this method is demonstrated by the rapid and biomimetic synthesis of clavicipitic acid.
Co-reporter:Yu-An Zhang, Qiang liu, Chao Wang, and Yanxing Jia
Organic Letters 2013 Volume 15(Issue 14) pp:3662-3665
Publication Date(Web):July 2, 2013
DOI:10.1021/ol401541t
A concise total synthesis of rugulovasine A is achieved by using Uhle’s ketone derivative as the key intermediate, which was synthesized by intramolecular cyclization via metal–halogen exchange. Two different routes to construct a spirocyclic butyrolactone subunit involving a Ru-catalyzed cyclocarbonylation and a special Ru-catalyzed double bond rearrangement were studied.
Co-reporter:Shiyong Mao, Yanxing Jia
Tetrahedron Letters 2013 Volume 54(Issue 32) pp:4343-4345
Publication Date(Web):7 August 2013
DOI:10.1016/j.tetlet.2013.06.027
Attempts to the synthesis of the A ring of halichomycin via ring-closing metathesis (RCM) reaction were investigated. When triene 5 was used as the precursor of cyclization, only unexpected byproduct aldehyde 17 was obtained. When diene 6 was used as the precursor of cyclization, the desired product 20 was obtained in reasonable yield. This work demonstrated that both modification of the substrate and the RCM reaction conditions are important for obtaining the desired 11-membered macrocycle in reasonable yield.
Co-reporter:Zhouting Rong, Qingjiang Li, Wenhan Lin, Yanxing Jia
Tetrahedron Letters 2013 Volume 54(Issue 33) pp:4432-4434
Publication Date(Web):14 August 2013
DOI:10.1016/j.tetlet.2013.06.028
The simple and one-pot method for the synthesis of polysubstituted tetrahydroquinolines from readily available anilines and aldehydes under reagent-free conditions has been developed. The scope of the transformation has been demonstrated. This method has been successfully applied to the rapid formal synthesis of (±)-martinellic acid and martinelline.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dong Shan;Yan Gao; Yanxing Jia
Angewandte Chemie 2013 Volume 125( Issue 18) pp:5002-5005
Publication Date(Web):
DOI:10.1002/ange.201300571
Co-reporter:Weimin Hu;Hua Qin; Yuxin Cui; Yanxing Jia
Chemistry - A European Journal 2013 Volume 19( Issue 9) pp:3139-3147
Publication Date(Web):
DOI:10.1002/chem.201204137
Abstract
In this article, the total syntheses of antimalarial compound decursivine and its biologically inactive sibling serotobenine are presented. The biomimetic synthesis of (±)-serotobenine was investigated first, but failed. During the subsequent investigation of other synthetic routes, we discovered a new cascade Witkop photocyclization/elimination/addition sequence, which enabled the expedient synthesis of not only racemic decursivine and serotobenine, but also enantiopure (+)- and (−)-decursivine and a variety of their analogues. The present syntheses represent the shortest pathway for the total synthesis of decursivine and serotobenine to date. Moreover, the newly developed cascade sequence for the total synthesis of decursivine does not need any protecting steps. The scope and the reaction mechanism of the cascade sequence were also studied. A rational mechanism for the cascade sequence is proposed, which is consistent with the previous studies and our current experimental results.
Co-reporter:Dong Shan;Yan Gao; Yanxing Jia
Angewandte Chemie International Edition 2013 Volume 52( Issue 18) pp:4902-4905
Publication Date(Web):
DOI:10.1002/anie.201300571
Co-reporter:Qiang Liu, Yu-An Zhang, Ping Xu, and Yanxing Jia
The Journal of Organic Chemistry 2013 Volume 78(Issue 21) pp:10885-10893
Publication Date(Web):October 10, 2013
DOI:10.1021/jo4018777
We report the enantioselective total synthesis of (+)-lysergic acid using two different strategies, which featured three metal-catalyzed reactions for the construction of the BCD three rings, involving Pd-catalyzed indole synthesis for the construction of the B ring, a ring-closing metathesis reaction for the formation of the D ring, and an intramolecular Heck reaction to forge the C ring. In synthetic strategy I, the synthesis was achieved in 20 steps following the ring construction sequence of BDC. In synthetic strategy II, the synthetic route was shortened to only 12 steps by following the ring construction sequence of DBC and using a 4-chlorotryptophan derivative for the intramolecular Heck reaction. Moreover, we also discussed an unsuccessful synthetic strategy.
Co-reporter:Jingjing Liang, Weimin Hu, Pengyu Tao, and Yanxing Jia
The Journal of Organic Chemistry 2013 Volume 78(Issue 11) pp:5810-5815
Publication Date(Web):May 13, 2013
DOI:10.1021/jo400841d
The concise synthesis of the novel telomerase inhibitors dictyodendrins B and E was completed in only 9 and 11 steps (longest linear sequence). The highly convergent strategy employed a palladium-catalyzed Larock indole synthesis and a palladium-mediated one-pot consecutive Buchwald–Hartwig amination/C–H activation reaction as key steps. The present synthesis exhibits respectable levels of atom-, redox-, and step-economy.
Co-reporter:Yonghua Liu, Liangren Zhang, Yanxing Jia
Tetrahedron Letters 2012 Volume 53(Issue 6) pp:684-687
Publication Date(Web):8 February 2012
DOI:10.1016/j.tetlet.2011.11.118
The total synthesis of maremycins A, B, C1/C2, D1, and D2 is achieved starting from the natural amino acids l-isoleucine and S-methyl-l-cysteine, in which the total synthesis of maremycins B, C1/C2, and D2 is accomplished for the first time. The synthesis features a position-selective intramolecular bromination process for the synthesis of key chiral building block, a Pd-catalyzed indole synthesis for the preparation of (2S,3S)-β-methyltryptophan and hydroxylation of oxindoles by molecular oxygen. In addition, the protocol for conversion of maremycins A and B to maremycins C and D was improved.
Co-reporter:Danfeng Jiang, Zhengren Xu, Yanxing Jia
Tetrahedron 2012 68(22) pp: 4225-4232
Publication Date(Web):
DOI:10.1016/j.tet.2012.03.097
Co-reporter:Qingjiang Li, Shiyong Mao, Yuxin Cui, and Yanxing Jia
The Journal of Organic Chemistry 2012 Volume 77(Issue 8) pp:4111-4116
Publication Date(Web):March 26, 2012
DOI:10.1021/jo202602n
An efficient and convergent synthesis of the C5–C18 fragment of halichomycin is reported. Butanolide fragment 6 was readily prepared stereoselectively from (R)-Roche ester through catalyst control; dienylic bromide domain 7 was synthesized from (S)-serine by substrate control. C5–C18 fragment 2 was rapidly assembled through a stereoselective alkylation of the butanolide with the dienylic bromide, followed by functional group transformations.
Co-reporter:Qiang Liu and Yanxing Jia
Organic Letters 2011 Volume 13(Issue 18) pp:4810-4813
Publication Date(Web):August 25, 2011
DOI:10.1021/ol2018467
A stereocontrolled total synthesis of (+)-lysergic acid (1) is achieved using three metal-catalyzed methodologies for the construction of three key rings. Highlights of the synthesis include Pd-catalyzed indole synthesis to form the B ring, a RCM reaction to form the D ring, and an intramolecular Heck reaction to form the C ring.
Co-reporter:Zhengren Xu, Fengying Zhang, Lihe Zhang and Yanxing Jia
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 7) pp:2512-2517
Publication Date(Web):06 Jan 2011
DOI:10.1039/C0OB01115K
The total synthesis of protein kinase C activator (−)-indolactam V (IL-V) has been successfully completed with two separate approaches: From known 4-nitrotryptophan derivative 3 in 8 steps (49% overall yield) and from L-glutamic acid in 12 steps (18% overall yield), where 4-nitrotryptophanol derivative 4 served as a key intermediate. Derivatives 3 and 4, both incorporating indole 4-substitution and the C-9 stereocenter in IL-V, were synthesized via the Pd-catalyzed indole synthesis from 3-nitro-2-iodoaniline 5 with aldehydes 6 and 7, respectively. Aldehyde 7 was, meanwhile, synthesized from L-glutamic acid in 5 steps (68% yield). Lactamization of the 9-membered ring was achieved using HATU in THF in good yield.
Co-reporter:Hua Qin;Zhengren Xu; Yuxin Cui; Yanxing Jia
Angewandte Chemie 2011 Volume 123( Issue 19) pp:4539-4541
Publication Date(Web):
DOI:10.1002/ange.201100495
Co-reporter:Hua Qin;Zhengren Xu; Yuxin Cui; Yanxing Jia
Angewandte Chemie International Edition 2011 Volume 50( Issue 19) pp:4447-4449
Publication Date(Web):
DOI:10.1002/anie.201100495
Co-reporter:Zhiyao Lu, Chunmei Hu, Jiajie Guo, Jing Li, Yuxin Cui and Yanxing Jia
Organic Letters 2010 Volume 12(Issue 3) pp:480-483
Publication Date(Web):December 23, 2009
DOI:10.1021/ol902672a
A palladium-catalyzed domino reaction involving a C−H activation process to synthesize diverse carbo- and heterocyclic skeletons was developed. H2O (as cosolvent) is used to control the regioselectivity.
Co-reporter:Weimin Hu, Fengying Zhang, Zhengren Xu, Qiang Liu, Yuxin Cui and Yanxing Jia
Organic Letters 2010 Volume 12(Issue 5) pp:956-959
Publication Date(Web):January 28, 2010
DOI:10.1021/ol902944f
A highly stereocontrolled and efficient total synthesis of (−)-stephanotic acid methyl ester and (−)-celogentin C was accomplished in longest linear 14 steps (4.6% overall yield) and in 20 steps (1.6% overall yield) from l-tryptophan, respectively. Highlights of the synthesis include a tandem asymmetric Michael addition/bromination/azidation strategy for a ready access to the leucine-tryptophan moiety (Leu-Trp linkage) and an oxidative coupling reaction to form the indole-imidazole linkage.
Co-reporter:Qingjiang Li, Aili Fan, Zhiyao Lu, Yuxin Cui, Wenhan Lin, and Yanxing Jia
Organic Letters 2010 Volume 12(Issue 18) pp:4066-4069
Publication Date(Web):August 24, 2010
DOI:10.1021/ol101644g
A simple and efficient method for the synthesis of 1,3,4-trisubstituted or 3,4-disubstituted pyrroles has been developed. The reaction represents the first time that pyrroles are synthesized directly from readily available aldehydes and amines (anilines) as starting materials. This method has been successfully applied to the rapid synthesis of purpurone.
Co-reporter:Jing Li, Yong Jiang, Qingjiang Li, Qiang Xiao, Yanxing Jia, Pengfei Tu
Tetrahedron Letters 2010 Volume 51(Issue 7) pp:1121-1123
Publication Date(Web):17 February 2010
DOI:10.1016/j.tetlet.2009.12.118
A formal synthesis of semiaquilegin A is achieved starting from readily available oridonin in 19 linear steps. The absolute configuration of the natural product has been established. A variety of useful analogues were prepared through this synthetic route.
Co-reporter:Chao Li;Chao Du;Hua Tian;Chao Jiang;Min Du;Yan Liu; Ren-Zhong Qiao; Yan-Xing Jia; Yu-Fen Zhao
Chemistry - A European Journal 2010 Volume 16( Issue 43) pp:12935-12940
Publication Date(Web):
DOI:10.1002/chem.201000552
Abstract
A new family of artificial transcription factor (ATF)-based conjugates have been designed and synthesized as potent chemical nucleases. Polyamides as the important and efficient ATFs were used to modify and activate several anchor compounds. The results demonstrate that the resulting conjugates remarkably promote the rate accelerations and non-random double-strand DNA cleavage activity. Interestingly, the cleavage activity of both the hydrolytic and oxidative agents was promoted efficiently through the modification of the ATFs.
Co-reporter:Chunmei Hu, Hua Qin, Yuxin Cui, Yanxing Jia
Tetrahedron 2009 65(45) pp: 9075-9080
Publication Date(Web):
DOI:10.1016/j.tet.2009.09.050
Co-reporter:Fengying Zhang, Yanxing Jia
Tetrahedron 2009 65(34) pp: 6840-6843
Publication Date(Web):
DOI:10.1016/j.tet.2009.06.068
Co-reporter:Pengyu Tao and Yanxing Jia
Chemical Communications 2014 - vol. 50(Issue 55) pp:NaN7370-7370
Publication Date(Web):2014/05/21
DOI:10.1039/C4CC02947J
The rhodium(III)-catalyzed intramolecular annulation of alkyne-tethered acetanilides for the synthesis of fused tricyclic indole scaffolds via C–H activation has been developed, which has the potential for the synthesis of many indole alkaloids. This reaction proceeds under mild reaction conditions and with tolerance to a variety of functional groups.
Co-reporter:Zhengren Xu, Fengying Zhang, Lihe Zhang and Yanxing Jia
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 7) pp:NaN2517-2517
Publication Date(Web):2011/01/06
DOI:10.1039/C0OB01115K
The total synthesis of protein kinase C activator (−)-indolactam V (IL-V) has been successfully completed with two separate approaches: From known 4-nitrotryptophan derivative 3 in 8 steps (49% overall yield) and from L-glutamic acid in 12 steps (18% overall yield), where 4-nitrotryptophanol derivative 4 served as a key intermediate. Derivatives 3 and 4, both incorporating indole 4-substitution and the C-9 stereocenter in IL-V, were synthesized via the Pd-catalyzed indole synthesis from 3-nitro-2-iodoaniline 5 with aldehydes 6 and 7, respectively. Aldehyde 7 was, meanwhile, synthesized from L-glutamic acid in 5 steps (68% yield). Lactamization of the 9-membered ring was achieved using HATU in THF in good yield.
Co-reporter:Lei Guo, Fengying Zhang, Weimin Hu, Lei Li and Yanxing Jia
Chemical Communications 2014 - vol. 50(Issue 25) pp:NaN3302-3302
Publication Date(Web):2014/02/07
DOI:10.1039/C3CC49717H
A new palladium-catalyzed method for the synthesis of benzofurans by reaction of 2-hydroxystyrenes and iodobenzenes via a C–H activation/oxidation tandem reaction has been unprecedentedly discovered. By using this strategy, the overall synthetic efficiency of the synthesis of decursivine and its analogues was indeed improved. Preliminary mechanistic studies shed light into the possible mechanisms.
Co-reporter:Xiwu Zhang, Haichao Liu and Yanxing Jia
Chemical Communications 2016 - vol. 52(Issue 49) pp:NaN7667-7667
Publication Date(Web):2016/05/19
DOI:10.1039/C6CC02600A
A concise and efficient synthetic route to 4,5-fused tricyclic 2-quinolones through the palladium-catalyzed carbonylative annulation of alkyne-tethered N-substituted o-iodoanilines has been developed. This reaction proceeds smoothly under mild reaction conditions and exhibits exceptional tolerance to a variety of functional groups. It has been successfully applied to the efficient synthesis of BI 224436, an HIV integrase inhibitor.
Co-reporter:Yinliang Guo, Qiang Liu and Yanxing Jia
Chemical Communications 2015 - vol. 51(Issue 5) pp:NaN891-891
Publication Date(Web):2014/11/24
DOI:10.1039/C3CC48897G
A concise synthesis of the tricyclic skeleton of crotobarin and crotogoudin via a gold-catalyzed 1,6-enyne cycloisomerization reaction is reported.
Co-reporter:Pengyu Tao, Zhuang Chen and Yanxing Jia
Chemical Communications 2016 - vol. 52(Issue 75) pp:NaN11303-11303
Publication Date(Web):2016/08/25
DOI:10.1039/C6CC05930A
The enantioselective total synthesis of the 3,4-fused indole alkaloid ht-13-A has been achieved. The synthesis features a zinc-mediated propargylation, a rhodium-catalyzed intramolecular C–H activation reaction, and a methylamine-mediated cyclization. This concise and efficient synthetic approach can be applied for the gram-scale synthesis of ht-13-A.



](http://img.cochemist.com/ccimg/59800/59738-27-1.png)
](http://img.cochemist.com/ccimg/59800/59738-27-1_b.png)





















![Benzenemethanol, 2-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/405200/405198-79-0.png)
![Benzenemethanol, 2-[(4-methoxyphenyl)ethynyl]-](http://img.cochemist.com/ccimg/405200/405198-79-0_b.png)


![Benzenemethanol, 2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/229100/229027-92-3.png)
![Benzenemethanol, 2-[(trimethylsilyl)ethynyl]-](http://img.cochemist.com/ccimg/229100/229027-92-3_b.png)

![5,9-Methano-2H-cyclohepta[b]furo[2,3-d]furan-2,6(3H)-dione,8-ethenyl-3-ethylideneoctahydro-4a,7,7,9-tetramethyl-,(3E,3aS,4aR,5S,8R,9S,9aS,9bS)-](http://img.cochemist.com/ccimg/156300/156250-64-5.png)
![5,9-Methano-2H-cyclohepta[b]furo[2,3-d]furan-2,6(3H)-dione,8-ethenyl-3-ethylideneoctahydro-4a,7,7,9-tetramethyl-,(3E,3aS,4aR,5S,8R,9S,9aS,9bS)-](http://img.cochemist.com/ccimg/156300/156250-64-5_b.png)
![1,3,4,5-tetrahydro-8-methoxy-5-methyl-Pyrrolo[4,3,2-de]quinoline](http://img.cochemist.com/ccimg/154500/154490-02-5.png)
![1,3,4,5-tetrahydro-8-methoxy-5-methyl-Pyrrolo[4,3,2-de]quinoline](http://img.cochemist.com/ccimg/154500/154490-02-5_b.png)
![2,4,7-trimethyloctahydro-1H-cyclopenta[c]pyridin-6-ol](http://img.cochemist.com/ccimg/145400/145307-22-8.png)
![2,4,7-trimethyloctahydro-1H-cyclopenta[c]pyridin-6-ol](http://img.cochemist.com/ccimg/145400/145307-22-8_b.png)
![2(1H)-Pyrazinone,6-[4-[1-(2-amino-2,3-dihydro-1H-imidazol-4-yl)ethyl]-7-hydroxy-1H-indol-3-yl]-3-(6-bromo-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/143000/142979-34-8.png)
![2(1H)-Pyrazinone,6-[4-[1-(2-amino-2,3-dihydro-1H-imidazol-4-yl)ethyl]-7-hydroxy-1H-indol-3-yl]-3-(6-bromo-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/143000/142979-34-8_b.png)






![Benzaldehyde, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/116800/116748-05-1.png)
![Benzaldehyde, 4-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-](http://img.cochemist.com/ccimg/116800/116748-05-1_b.png)

![Lithium, [5-(trimethylsilyl)-2-furanyl]-](http://img.cochemist.com/ccimg/114200/114173-77-2.png)
![Lithium, [5-(trimethylsilyl)-2-furanyl]-](http://img.cochemist.com/ccimg/114200/114173-77-2_b.png)
![Phenol, 2-[1-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/110600/110598-51-1.png)
![Phenol, 2-[1-(4-methoxyphenyl)ethenyl]-](http://img.cochemist.com/ccimg/110600/110598-51-1_b.png)
![Indolo[1,2-a][1,8]naphthyridine-7-ethanol,4a-ethyl-1,2,3,4,4a,5,6,12a-octahydro-, (4aR,12aS)-](http://img.cochemist.com/ccimg/109800/109794-95-8.png)
![Indolo[1,2-a][1,8]naphthyridine-7-ethanol,4a-ethyl-1,2,3,4,4a,5,6,12a-octahydro-, (4aR,12aS)-](http://img.cochemist.com/ccimg/109800/109794-95-8_b.png)








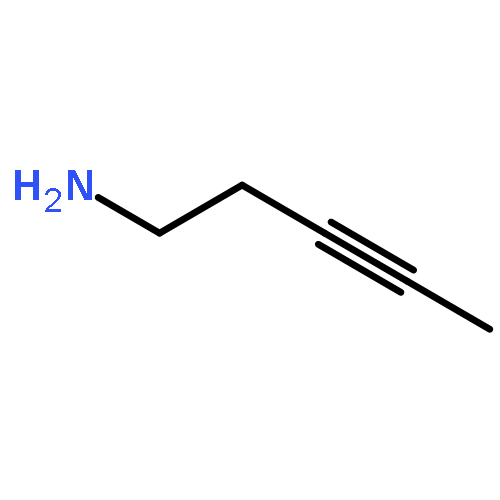
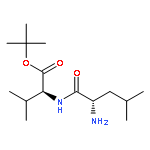
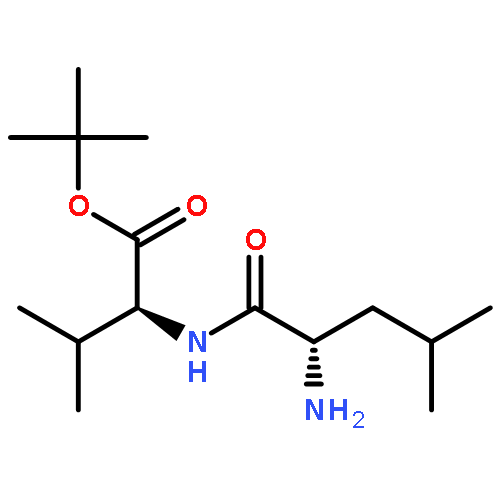
![2-CYCLOHEXEN-1-ONE, 5-[1-(CHLOROMETHYL)ETHENYL]-2-METHYL-, (R)-](http://img.cochemist.com/ccimg/86900/86847-23-6.png)
![2-CYCLOHEXEN-1-ONE, 5-[1-(CHLOROMETHYL)ETHENYL]-2-METHYL-, (R)-](http://img.cochemist.com/ccimg/86900/86847-23-6_b.png)


![[1,1'-Biphenyl]-2-amine, 4',6-dimethoxy-](http://img.cochemist.com/ccimg/84300/84251-38-7.png)
![[1,1'-Biphenyl]-2-amine, 4',6-dimethoxy-](http://img.cochemist.com/ccimg/84300/84251-38-7_b.png)